
Case Report
Austin J Urol. 2015;2(2): 1023.
Primary Hyperoxaluria and Renal Cortical Nephrocalcinosis – as a Cause of Renal Failure
Sriram K¹*, Ramesh Chinnasamy², Prabha Senguttavan² and Shroff S¹
¹Department of Urology, Sri Ramachandra University, India
²Department of Nephrology, Sri Ramachandra University, India
*Corresponding author: Sriram K, Department of Urology, Sri Ramachandra University, Ramachandra Nagar, Porur, Chennai, India
Received: January 27, 2015; Accepted: February 25, 2015; Published: March 03, 2015
Abstract
A nine year old male child presented with end stage renal disease, needing emergency haemodialysis. During evaluation, he was diagnosed to have primary hyperoxaluria. The need for transplantation and the problems associated with an isolated renal transplantation were discussed with the patient. His parents subsequently opted for continuous ambulatory peritoneal dialysis.
The Primary Hyperoxalurias (PH) are inborn errors of metabolism of Glyoxylate and oxalate resulting in an increase in the endogenous production of oxalate that in turn results in an excessive excretion of oxalates in urine.
As a result of a high urinary oxalate excretion, the urine becomes supersaturated with calcium oxalate crystals, resulting in formation of crystals within the tubular lumen. PH therefore, manifests either as severe Nephrocalcinosis and/or urolithiasis.
Transplantation in these patients with renal failure has to be done with extreme caution. It is sensible to combine Renal and liver transplantation in order to prevent recurrence of symptoms.
The latency time between the presentation of symptoms and the time a diagnosis is made is too wide and protracted. Hence it is imperative to have a high index of suspicion in making a diagnosis of hyperoxaluria. Early diagnosis and appropriate treatment are obligatory to prevent this potentially devastating clinical condition.
Keywords: Hyperoxaluria, Renal transplantation, Nephrocalcinosis
Introduction
Primary Hyperoxaluria (PH) is an inborn error of metabolism, where there is an excessive production and urinary excretion of oxalates. The natural history of PH is such that this problem presents early in childhood as calcium oxalate stones and nephrocalcinosis with recurrent urinary tract infection and progresses with increase in time to full blown renal failure and death. The main hallmark of this particular disease is that there are no obvious biochemical abnormalities, except an increased urinary oxalate excretion [1]. In most of the patients, the disease takes a fatal course, as they develop terminal renal failure before the end of third decade of life [2]. Demonstration of an increased urinary oxalate excretion is necessary to distinguish this condition from Calcium oxalate urolithiasis without hyperoxaluria.
Case Report
A nine year old male child presented with fatiguability, recurrent episodes of vague bilateral loin pain, puffiness of face, pedal edema and recurrent vomiting episodes.
On evaluation, he was diagnosed to have abnormal renal function. Ultrasound abdomen revealed parenchymal calcifications of both the kidneys. The serum creatinine level was 11.8 mg/dl and serum potassium was 5.6 mEq/L. The serum uric acid levels were 12 mg/ dl; serum calcium was 7 mg/dl. He initially underwent haemodialysis through right jugular access and was subsequently worked up for a possible renal transplantation on a later date.
The 24 hour urine volume was about 800 ml on an average. The urinary excretion of calcium was 180mg/24 hours; uric acid excretion of 480 mg per 24 hours and phosphates being 450 mg. The urinary oxalate levels were 148 mg per 24 hours. Plain Computed Tomography (CT) scan of the abdomen revealed an extensive parenchymal calcification involving both the kidneys. The upper calyx of the right kidney also harbored a 2 centimeter sized stone occupying the entire dilated upper calyx, extending onto and occluding the infundibulum of the right upper calyx, measuring 1200 Houndsfield (HU) units. The pelvi-calyceal system and the ureters were not dilated.
Based on the available clinical and biochemical findings, a diagnosis of primary hyperoxaluria was considered. The diagnosis of hyperoxaluria needs either a liver biopsy to measure the AGT catalytic activity or genetic analysis to study the gene mutations. As these facilities were not available in our centre, based on the clinical, radiological and biochemical findings, a diagnosis of primary hyperoxaluria was made. The condition of the patient was discussed in detail with his parents and also the need for combined liver and renal transplantation and a very high possibility of disease recurrence after isolated renal transplantation. The patient subsequently opted for continuous ambulatory peritoneal dialysis.
Discussion
Renal cortical nephrocalcinosis is one of the rare conditions encountered in urological practice. Calcifications in the kidney may either be within the renal parenchyma or within the collecting system. The former is called as Nephrocalcinosis, which may either be cortical or medullary depending on the location of the calcification, and the latter is termed Nephrolithiasis [3].
The primary hyperoxalurias are inborn errors of metabolism of Glyoxylate and oxalate resulting in an increase in the endogenous production of oxalate that in turn results in an excessive excretion of oxalates in urine.
Owing to a high urinary oxalate excretion, the urine becomes supersaturated with calcium oxalate crystals, resulting in formation of crystals within the tubular lumen. PH therefore, manifests either as severe Nephrocalcinosis and/or urolithiasis.
Progressive renal parenchymal inflammation and interstitial fibrosis due to Nephrocalcinosis cause renal impairment, which progresses to an end stage renal disease status over a period of time [4].
The various causes of hyperoxaluria are:
Primary hyperoxaluria -- disorders in biosynthetic pathways.
Enteric hyperoxaluria – intestinal malabsorptive states associated with inflammatory bowel disease, celiac sprue, or intestinal resection.
Dietary hyperoxaluria-- excessive dietary intake or high substrate levels vitamin C.
PH is a rare metabolic disorder, transmitted as an Autosomal recessive disease. The primary pathology is a defect of the peroxysmal hepatic enzyme L-alanine: Glyoxylate Amino Transferase (AGT), an enzyme that helps in the conversion of Glyoxylate to Glycine that in turn results in the increase of Glyoxylate pool, which gets converted to oxalate [5]. In these patients, both the kidneys show an extensive calcification of the renal parenchyma. (Figure 1 &2).
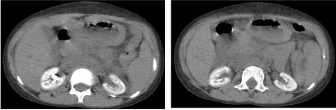
Figure 1: Cross sectional computed tomographyof the kidneys showing
extensive calcification of the parenchyma of both the kidney.
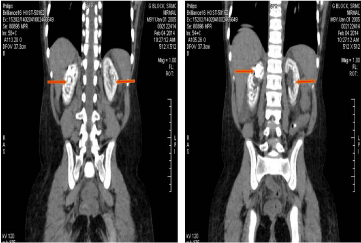
Figure 2: Coronal section of both kidneys, showing extensive calcification of
both renal parenchyma, with a stone in the right upper calyx.
There are three forms of PH, designated as types 1, 2 and 3 [6]. Type 1 is caused by a deficiency of the liver –specific enzyme, Alanine Glyoxylate Amino Transferase (AGT). Type 2 is caused by a lack of Glyoxylate reductase – hydroxyl pyruvate reductase. Type 3 results from defects in the liver specific mitochondrial enzyme 4-Hydroxy – 2-Oxoglutarate Aldolase (HOGA). Over production of oxalates is the feature that is common to all the types [7].
Type 1 disease is the most common and most devastating subtype that more often leads to end stage renal disease. Type 2 disease often has a less severe course. Type 3 disease has the least severe course and may be silent for a long period or may limit itself to stone formation. Nephrocalcinosis and renal failure are relatively uncommon in type 3 disease and systemic involvement has been very rare in this subtype [7].
An early and aggressive conservative treatment, including forced, high fluid intake supported by diuretic therapy, administration of calcium oxalate crystallization inhibitor and pyridoxine should be started as soon as the diagnosis of PH is made [8].
Transplantation in these patients with renal failure has to be done with extreme caution. Since liver is the major site of glyoxylate detoxification, the primary pathology of excess oxalate production would continue as long as the native liver exists. It is prudent to combine Renal and liver transplantation in order to prevent recurrence of symptoms [9].
Once the glomerular filtration rate falls below 30 million per 1.73 m2, decreased oxalate excretion by the kidneys, along with increased production of oxalates by the liver leads to an increase in the serum oxalate levels. When this exceeds the super saturation point of calcium oxalate, it results in a systemic deposition of oxalate, a term called as Oxalosis. Soft tissue calcification in various extra renal tissues is termed as Systemic Oxalosis. The various organs involved in Systemic oxalosis include Bone marrow, heart, nerves, joints, skin and retina [10]. Oxalate deposition in various tissues can lead to heart block, peripheral gangrene and crippling bone disease [11].
The median age at initial symptom of PH is 6 years in European population and median age at diagnosis is 7.3 years [12]. PH has a variable presentation. The infantile form is the most life threatening, due to a rapid progression to end stage renal disease status. Sonographic examination show either cortical or medullary nephrocalcinosis. Because of the relative infrequent incidence of the condition, the diagnosis is often delayed. Majority of the patients, at the time of diagnosis, had already reached end stage renal disease stage [13].
The time interval between the onset of symptoms and the time a diagnosis is arrived at is too wide and long. The purpose of this article is to drive home the message that there is a need for having a high index of suspicion in making a diagnosis of hyperoxaluria. In the absence of facility for liver biopsy or gene analysis, associated biochemical and radiological findings, nephrocalcinosis and renal impairment should be sufficient enough to make a diagnosis of hyperoxaluria [14] .
The diagnostic approaches need to be improved and sophisticated. It is advisable that every pediatric patient with nephrocalcinosis should undergo urinary oxalate estimation. Isolated renal transplantation, even though anecdotal successful reports are published in literature, should be advised with extreme caution [15].
Conclusions
All paediatric patients with calcium oxalate stones need to undergo a thorough metabolic evaluation. If left undiagnosed or untreated, the disease usually takes a fatal course. The ultimate goal in treating these patients is to prevent or delay the inevitable onset of renal failure. Early diagnosis and appropriate treatment are mandatory to prevent this devastating clinical condition.
References
- Archer HE, Dormer AE, Scowen EF, Watts RW. The aetiology of primary hyperoxaluria. Br Med J. 1958; 1: 175-181.
- O'Regan PF, Joekes AM. Primary hyperoxaluria. J R Soc Med. 1980; 73: 541-544.
- Schepens D, Verswijvel G, Kuypers D, Vanrenterghem Y. Images in Nephrology. Renal cortical nephrocalcinosis. Nephrol Dial Transplant. 2000; 15: 1080-1082.
- Hoppe B, Beck BB, Milliner DS. The primary hyperoxalurias. Kidney Int. 2009; 75: 1264-1271.
- Primary hyperoxaluria. Patrick Niaudet. Orphanet encyclopedia.
- Harambat J, Fargue S, Bacchetta J, Acquaviva C, Cochat P. Primary hyperoxaluria. Int J Nephrol. 2011; 2011: 864580.
- Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. 2013; 369: 649-658.
- Cochat P, Basmaison O. Current approaches to the management of primary hyperoxaluria. Arch Dis Child. 2000; 82: 470-473.
- Bergstralh EJ, Monico GC, Lieske CJ, Herges MR, Langman BC, Hoppe B, et al. Transplantation outcomes in primary hyperoxaluria.. American J transplant. 2010; 10: 2493-2501.
- Sriram K, Kekre NS, Gopalakrishnan G. Primary hyperoxaluria and systemic oxalosis. Indian J Urol. 2007; 23: 79-80.
- Woolfson RG, Mansell MA. Hyperoxaluria and renal calculi. Postgrad Med J. 1994; 70: 695-698.
- van Woerden CS, Groothoff JW, Wanders RJ, Davin JC, Wijburg FA. Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrol Dial Transplant. 2003; 18: 273-279.
- Hoppe B, Langman CB. A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatr Nephrol. 2003; 18: 986-991.
- Hoppe B, Leumann E. Diagnostic and therapeutic strategies in hyperoxaluria: a plea for early intervention. Nephrol Dial Transplant. 2004; 19: 39-42.
- O'Regan P, Constable AR, Joekes AM, Kasidas GP, Rose GA. Successful renal transplantation in primary hyperoxaluria. Postgrad Med J. 1980; 56: 288-293.