
Research Article
Austin J Public Health Epidemiol. 2019; 6(2): 1085.
Hepatitis C Outbreak in Haemato-Oncology Ward: A Challenge of Investigating the Transmission Mechanism in Patients with Multiple Exposures
Rosinska M1*, Stepien M1, Caraballo Cortes K3, Janiak M3, Radkowski M3, Godzik P1, Gorczyca A2, Wszolek M2, Orzel-Nowak A2, Olszowska-Pason R2, Grabarczyk P4 and Sadkowska-Todys M1
1National Institute of Public Health – National Institute of Hygiene, Poland
2State Sanitary Inspection, Poland
3Department of Immunopathology of Infectious and Parasitic Diseases, Medical University of Warsaw, Poland
4Department of Virology, Institute of Hematology and Transfusiology, Poland
*Corresponding author: Rosinska M, National Institute of Public Health – National Institute of Hygiene, Warsaw, Poland
Received: July 01, 2019; Accepted: July 29, 2019; Published: August 05, 2019
Abstract
Background: Nosocomial transmission of Hepatitis C Virus (HCV) continues to occur, even in developed countries. Recent HCV outbreaks most often concern vulnerable populations, such as patients of haemodialysis units, oncology wards and CT/MRI scanning units. We report an investigation of an outbreak in haemato - oncology ward to determine transmission mechanisms and to discuss challenges arising in these settings.
Methods: We include as cases previously undiagnosed HCV infected patients, hospitalized in the haemato-oncology ward between 1st August and 31st October with outbreak strain confirmed with Next-Generation Sequencing (NGS) analysis of Hypervariable Region 1 (HVR1). The required similarity threshold for outbreak strain was 3.7% genetic distance. We selected the exposure period based on HCV incubation time, due to multiple hospitalizations of all patients in the implicated ward. We attempted to screen all patients hospitalized during exposure period and collected exposure data from medical records.
Results: Of 129 people eligible for screening, 34 died before being reached, 17 refused or could not be contacted, and 78 were tested. HCV infection was confirmed (HCV-RNA) in 11 (14%) patients, of whom in seven HVR1 amplification was feasible and all harboured the outbreak strain. Reception of chemotherapy in August 16-31 (AOR 30.17, 95% CI 2.45-371.21) and in October 1-15 (35.09, 2.53-487.28) was significantly associated with infection. Infected batches were excluded as source since patients received different regimens. However, minor procedures, such as i.v. line flushing, were not fully documented. Multidose vials of saline were used.
Conclusion: Our results indicate a close relationship of the virus in the haemato-oncology ward patients suggesting a common source of infection, despite inconclusive exposure analysis. Plausible transmission route includes breaches in minor procedures. As HCV outbreak investigations inevitably rely on medical documentation, we recommend that at least those minor procedures, which were previously linked to transmission, be documented in detail.
Keywords: HCV; Hepatitis C; Healthcare associated infections; Infection transmission; Molecular epidemiology
Introduction
European surveillance data attribute the majority of newly diagnosed Hepatitis C (HCV) infections to injecting drug use. Likewise, people who inject drugs constitute the most affected group in the European countries [1–3]. Nonetheless, a substantial proportion of HCV cases do not report injecting drug use. While some of the cases, especially among men who have sex with men appear to be transmitted sexually, many report only exposures to medical invasive procedures. Although infrequent in developed countries, healthcare related transmission risk still persists despite elimination of the risk associated with blood transfusions [4,5] and it may be especially relevant to patients with conditions requiring frequent medical interventions, in populations with higher background prevalence such as patients of haemodialysis units or diabetes patients [4,6,7]. Infections occur in relation to breeches in safety procedures or implementation of inadequate procedures, notably during unsafe injections [4].
In Poland the surveillance data indicate that the past or current transmission related to medical procedures continues to impact the current hepatitis C burden. This is confirmed by seroprevalence and case-control studies identifying transfusion before 1992 as the key risk factor for prevalent cases. Other risk factors include multiple hospitalizations and minor medical procedures. The association with particular procedures is not consistent across studies, drawing attention to the fact that different medical procedures may be involved in different populations [8–10]. Furthermore, medical procedures, especially minor medical and dental procedures tend to still contribute to the spread of the HCV infection in Poland [11–13].
Occurrence of nosocomial outbreaks may be also considered as an indicator of recent transmission events, for which the actual transmission mechanism could be identified. In a review carried out in the United States a substantial number of HCV outbreaks was identified in outpatient settings, particularly in haemodialysis centres, but also in hematology/ oncology and pain remediation clinics [6]. Similar HCV outbreaks have been also reported in Europe. In the published literature, the most commonly reported hepatitis C outbreaks in Europe concern haemodialysis units, oncology wards and CT/MRI scanning units [14].
Outbreak investigations in case of HCV infection can pose different methodological challenges. Due to predominantly asymptomatic course of disease, it is very likely that many outbreaks, especially smaller ones, are not identified. Moreover, as cases are usually diagnosed with delay, the confirmation of a link between cases is not evident, unless genetic identification of the outbreak strains is performed. The genetic analysis is complicated by existence of multiple quasi-species within one host [15], given documented possibility of transmission of minority variants [16]. Next Generation Sequencing (NGS) - based variant analysis allows to resolve this difficulty [16,17].
We report an outbreak among haemato-oncology patients in a county hospital in Poland. The suspected outbreak was reported in December 2015 to local public health department, with five cases initially diagnosed among patients of haemato-oncology ward. In addition to investigation of the possible transmission mechanisms, we aim to discuss technical difficulties arising in these settings.
Methods
Initial information and the exposure period
At the time of the outbreak, report five patients (confirmed HCV-RNA) have already been diagnosed in the ward, two of whom developed jaundice and three tested due to high levels of ALT. The first patient was diagnosed in October, the second in November and the other three in December. Initially local clinicians suspected exacerbation of chronic HCV infection due to chemotherapy [18], which caused the delay in reporting of the outbreak. The possibility of reactivation was later excluded as the patients did not receive the implicated chemotherapy regimens and earlier HCV infection was not documented in any of the five patients [19]. The review of surveillance data at local and regional level identified additional two cases of acute hepatitis C (both were diagnosed because of jaundice) that reported hospitalization in the implicated ward, notified in December 2015 and January 2016. They also negated other major HCV exposures (injecting drugs, tattooing), although one patient also reported hospitalization in a different hospital. In order to identify the possible exposure time we aligned the hospitalization times of all patients. All of them were admitted in the implicated ward multiple times in 2015 and were hospitalized in August 2015, but no single day could be identified when all were present. Moreover, August would fall out of the typical incubation period for acute hepatitis C (3-12 weeks, on average 7 weeks) [20]. We considered, that there could be more than one transmission event or some of the cases could be unrelated to the outbreak. In addition, longer incubation time due to underlying disease or chemotherapy could be taken into account. It was shown that time to HCV seroconversion is longer in patients with haematological disorders [21,22].
Initially, we selected the presumed exposure period based on HCV incubation time and extended it to account for the period when they were all hospitalized. Finally, the exposure period encompassed the time between August 1st and October 31st.
Case definitions
The initial case definition for screening purposes was a previously undiagnosed HCV infected (confirmed by HCV-RNA test) patient, hospitalized in the haemato-oncology ward during the specified exposure period (August 1st and October 31st 2015).
The case definition used for analysis in addition included the results Next-Generation Sequencing (NGS) analysis of Hypervariable Region 1 (HVR1). The case was classified as probable if there was no sample available for genetic analysis or the HCV strain could not be isolated. A patient meeting the screening case definition was considered to be a confirmed outbreak case if at least one of the strains isolated from this individual had a genetic distance of less than 3.7% from at least one variant strain from another patient, i.e. the minimum pairwise distance was less than 3.7%. This criterion was suggested in prior review work [16].
We further supported the selection of the cases possibly from a single transmission event by comparing genetic distances observed between the control sequences and between the case and control sequences.
Data and sample collection
We screened all patients hospitalized during specified exposure period and collected exposure data from medical records. The patients were invited to take part in the investigation, when returning to the hospital for next cycles of chemotherapy or control visits. The remaining patients were contacted at their home address. The patients were offered HCV screening, and a venous blood sample was collected from those who gave their consent. The samples were processed in the hospital laboratory and the sera were frozen and shipped to National Institute of Public Health – National Institute of Hygiene and the Warsaw Medical University for testing. The medical record of the patients were reviewed for all medical procedures associated with possible percutaneous exposure that took place during the exposure period. Based on these data a questionnaire to extract data from other patients records was constructed. All cases were also interviewed with routine surveillance questionnaire comprising also life-style exposures.
We used 10 samples from unrelated acute hepatitis C identified in blood donation service as control samples for molecular characteristics of the HVR1 region of the virus.
Laboratory methods
Next-Generation Sequencing (NGS) analysis of Hypervariable Region 1 (HVR1) was used to search for relatedness of HCV variants as described in [23].
In brief, total RNA was extracted from 250 μl of serum using Trizol (Life Technologies, Carlsbad, CA, USA) and subjected to reverse transcription using AccuScript High Fidelity Reverse Transcriptase (Agilent Technologies, Santa Clara, CA, USA) and random hexamers. A region of 175 nt length encompassing HVR1 was amplified in two-step PCR using FastStart High Fidelity Taq DNA Polymerase (Roche, Indianapolis, IN, USA) using sequence-specific promers. Primers employed in the second PCR contained tags recognized by GS Junior sequencing platform, standard 10-nucleotide multiplex identifiers and target-complementary sequence. Approximately 3×107 DNA amplicons were subjected to emulsion PCR using the GS Junior Titanium emPCR Lib-A Kit (454 Life Sciences, Branford, CT, USA). Amplicons were sequenced according to the manufacturer’s protocol using GS Junior platform (454 Life Sciences). HVR1 variants were reconstructed using the program diri_sampler from the Shorah software suite [24]. Subsequently, reconstructed haplotypes of frequency >0.5% were aligned by MEGA (Molecular Evolutionary Genetics Analysis), version 6.0 (https://www.megasoftware.net/) [25].
Statistical analysis
Fisher exact test was used to compare distribution of categorical covariates between cases and controls. Medians of numerical variable were compared with Mann-Whitney test. We used logistic regression to estimate the odds ratio of being a confirmed outbreak case as compared to HCV-RNA negative patients. Due to small sample size we were not able to study the full multivariate model. We investigated the predictors important in univariable analysis in pairs to identify the strongest ones. For factors (exposures) perfectly predicting the outcome the separate indicator variables were created to describe whether the exposure occurred in August, in September or in October.
Results
Of 129 people eligible for screening, 34 died before being reached, 17 refused or could not be contacted, and 78 were tested. HCV infection was confirmed in 11 (14%) patients, of whom in seven HVR1 amplification was feasible.
NGS results and case classification
NGS analysis revealed intrahost variability of HVR1 both in the samples from examined patients and the control samples from acute HCV infections identified in blood donors. Among the seven patients the predominant strain accounted for 54.1% - 100% of frequency of all strains identified and among the controls the predominant strain accounted for 32.6%-97.6% of frequency. The pairwise minimal distances between the cases meeting the screening definition varied between 0.0% and 1.3%, while between patients and controls they remained between 9.7% - 21.8% (apart from one 0.0% distance, between Pt_8 and C_118) and between controls – 0.0% - 16.6% (Table 1). The minimal distances between the strains isolated from the patients coincided with the distances between the predominating strains. The patient Pt_8 harboured three unrelated strains, one of which was similar to the strains isolated from other cases (minimal pairwise distances from 0.0% to 1.3%). Another strain, with relative frequency of 3%, was closely related (0.0% distance) to the strains isolated from the control C_118. The minimal distances between the strains isolated from controls related to minority strains. Distances between the predominating strains exceeded 10%.
![]()
Minimum pairwise distance
Patient code
Patient code
Pt_02
Pt_03
Pt_04
Pt_05
Pt_06
Pt_07
Pt_08
C_11
C_14
C_16
C_52
C_56
C_93
C_110
C_114
C_118
Pt_03
0,6%
Pt_04
0,0%
0,6%
Pt_05
0,0%
0,6%
0,0%
Pt_06
0,6%
1,3%
0,6%
0,0%
Pt_07
0,6%
1,3%
0,6%
0,0%
0,0%
Pt_08
0,6%
1,3%
0,6%
0,0%
0,0%
0,0%
C_11
17,4%
18,2%
17,3%
16,7%
18,2%
18,2%
12,7%
C_14
17,4%
17,7%
17,3%
16,7%
18,2%
18,2%
9,7%
0,0%
C_16
17,8%
17,8%
16,8%
17,0%
18,6%
18,6%
11,9%
0,0%
0,0%
C_52
18,5%
18,5%
18,4%
17,7%
19,3%
19,3%
12,6%
11,3%
10,3%
0,0%
C_56
20,2%
20,2%
20,2%
19,4%
20,2%
20,2%
13,6%
14,5%
12,7%
15,8%
15,0%
C_93
17,4%
17,4%
16,8%
16,7%
18,2%
18,2%
9,7%
0,0%
0,0%
0,0%
9,7%
12,7%
C_110
18,3%
19,1%
18,3%
17,5%
19,1%
19,1%
15,2%
13,4%
12,7%
14,3%
15,0%
14,6%
12,7%
C_114
20,9%
20,9%
20,9%
20,1%
21,8%
21,8%
14,3%
12,1%
12,1%
16,6%
15,8%
11,2%
12,6%
16,1%
C_118
16,1%
15,3%
15,2%
15,3%
15,3%
15,3%
0,0%
12,0%
11,0%
11,1%
12,6%
13,6%
11,0%
16,1%
13,5%
C_120
19,3%
20,1%
19,1%
18,5%
18,5%
18,5%
16,0%
11,1%
11,1%
13,5%
13,5%
17,7%
11,1%
13,5%
16,0%
15,2%
The number of base substitutions per site from between sequences are shown. Analyses were conducted using the Maximum Composite Likelihood model [40]. The analysis involved 77 nucleotide sequences. Codon positions included were 1st+2nd+3rd+ Noncoding. All positions containing gaps and missing data were eliminated. There were a total of 155 positions in the final dataset. Evolutionary analyses were conducted in MEGA5 [25].
Table 1: Minimal pairwise distance between viral strains isolated from patients and from controls.
Based on this analysis the seven patients will be considered confirmed outbreak cases with possible single source infection. The four patients either for whom no samples were available or no amplification was achieved were classified as probable cases.
Patient characteristics
The proportion of females among the uninfected individuals, all HCV-RNA positive cases and confirmed outbreak cases was, 64.2% (43/67), 45% (5/11) and 42.9% (3/7), respectively, and the mean (median) age in years – 64.9 (67), 60.7 (64) and 64.7 (66), respectively. The most common clinical diagnosis related to hospitalization were lymphoma (20.5%), multiple myeloma (20.5%) and leukemia (17.9%). Other diagnoses included thrombocytopenia, anaemia, polyglobulia, myelodysplasia, monoclonal gammopathy. The distribution of main diagnoses was not significantly different between cases and controls (p=0.323).
Altogether four patients developed jaundice and only two seroconverted at the time of sample collection (Pt_3, Pt_9). In case of one patient (Pt_11) clinical history and the sample were not available. All patients were hospitalized multiple times during the presumed exposure period (Figure 1,2).
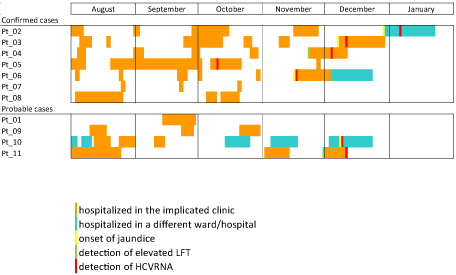
Figure 1: Summary of the exposure in the health care and HCV diagnosis among confirmed and probable outbreak case 1.
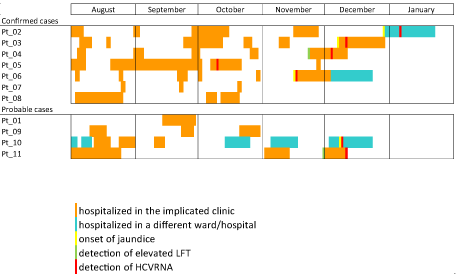
Figure 2: Summary of the exposure in the health care and HCV diagnosis among confirmed and probable outbreak case 2.
Exposure analysis
The overall hospitalization time during the exposure period (August – October 2015) was significantly longer among cases than among the uninfected patients hospitalized during the exposure period. Consequently, cases were more likely to be hospitalised in each of the examined months (Table 2). Significantly more cases than controls received chemotherapy, or blood transfusion or were subjected to a procedure outside of the implicated ward. Further investigation revealed that cases had different blood groups, excluding blood transfusion as a likely source of infection. Moreover, the category “procedures outside of the implicated ward” included different procedures performed in different medical facilities.
![]()
HCVRNA negative controls
Probable and confirmed cases
Confirmed cases
N(%)
N(%)
N(%)
Total
Total
67 (100.0)
10 (100.0)
7 (100.0)
Hospitalised in Aug 2015
No
28 (41.8)
1 (10.0)
0 (0.0)*
Yes
39 (58.2)
9 (90.0)
7 (100.0)
Hospitalised in Sep 2015
No
41 (61.2)
3 (30.0)
2 (28.6)
Yes
26 (38.8)
7 (70.0)
5 (71.4)
Hospitalised in Oct 2015
No
52 (77.6)
3 (30.0)**
1 (14.3)**
Yes
15 (22.4)
7 (70.0)
6 (85.7)
Received chemotherapy a biopsy in Aug-Oct 2015
No
43 (64.2)
3 (27.3)*
0 (0.0)**
Yes
24 (35.8)
8 (72.7)
7 (100.0)
Underwent a biopsy in Aug-Oct 2015
No
51 (76.1)
8 (72.7)
6 (85.7)
Yes
16 (23.9)
3 (27.3)
1 (14.3)
Underwent a trepan biopsy in Aug-Oct 2015
No
59 (88.1)
11 (100.0)
7 (100.0)
Yes
8 (11.9)
0 (0.0)
0 (0.0)
Underwent CT/MR scan with contrast in Aug-Oct 2015
No
58 (86.6)
9 (81.8)
6 (85.7)
Yes
9 (13.4)
2 (18.2)
1 (14.3)
Had a transfusion in Aug-Oct 2015
No
60 (89.6)
5 (45.5)**
4 (57.1)*
Yes
7 (10.4)
6 (54.5)
3 (42.9)
I.V. line placement in Aug-Oct 2015
No
8 (11.9)
0 (0.0)
0 (0.0)
Yes
59 (88.1)
11 (100.0)
7 (100.0)
Subcutaneous fraxiparine administration in Aug-Oct 2015
No
55 (82.1)
9 (81.8)
5 (71.4)
Yes
12 (17.9)
2 (18.2)
2 (28.6)
Catheter placement in Aug-Oct 2015
No
67 (100.0)
10 (90.9)
6 (85.7)
Yes
0 (0.0)
1 (9.1)
1 (14.3)
Insulin administration by pen in Aug-Oct 2015
No
64 (95.5)
11 (100.0)
7 (100.0)
Yes
3 (4.5)
0 (0.0)
0 (0.0)
Procedures outside of the implicated ward in Aug-Oct 2015
No
61 (91.0)
7 (63.6)*
4 (57.1)*
Yes
6 (9.0)
4 (36.4)
3 (42.9)
Total hospitalization length in Aug-Oct 2015 in days
mean (SD); median [range]
5.8 (0.6); 4 [1-29]
23.3 (4.9); 23 [6-65]***
25.4 (7.4); 23 [6-65]***
Table 2: Distribution of medical exposures among HCVRNA negative controls, probable and confirmed and confirmed cases only.
We further investigated the length of hospital stay and receiving the chemotherapy as the main risk factors for infection. We analysed the total lengths of hospital stay as well as the number of days spent in the implicated ward in August, September and October. All cases were hospitalized in August so the number of days in August was split into number of days in the August 1 – 15 and in August 16 – 31 to avoid perfect prediction problem. Similarly, all cases received chemotherapy and this variable was split into chemotherapy in August 1 -15, chemotherapy in August 15 -31, in September and in October. Increasing length of hospitalization predicted increased odds of infection (Table 3). When investigating the length of stay in August, September and October as separate variables in one model the length of stay in September was not significant, however both the number of hospitalization days in August and in October were significantly associated with HCV infection with an outbreak strain (AOR for each additional day in August 1.3, 95% CI 1.07-1.58 and for a day in October – 1.36, 95% CI 1.09-1.7). Receiving chemotherapy in either of the analysed time periods increased odds of infection, with the largest effect sizes for the August 16-31 and October, which was also split to October 1-15 and October 16-31 (Table 3). When analysing these variables in pairs, two significant independent effects emerged – reception of chemotherapy in August 16-31 (AOR 30.17, 95% CI 2.45-371.21) and in October 1-15 (35.09, 2.53-487.28).
![]()
OR*
95% CI
p-value
AOR**
95% CI
p-value
Duration of hospitalization in the implicated ward
Total number of days Aug - Oct
1.23
1.09-1.39
0.001
Number of days Aug
1.28
1.08-1.52
0.004
1.30#
1.07-1.58
0.008
Number of days Sep
1.17
1.01-1.35
0.033
Number of days Oct
1.31
1.12-1.54
0.001
1.36#
1.09-1.7
0.006
Reception of chemotherapy
Aug 1-15
5.05
1.01-25.22
0.049
Aug 16-31
44.25
4.7-416.53
0.001
30.17##
2.45-371.21
0.008
Sep
9.83
1.85-52.19
0.007
Oct
31
4.75-202.34
<0.001
Oct 1-15
39.38
5.74-270.25
<0.001
35.09##
2.53-487.28
0.008
Oct 16-31
28.44
4.29-188.75
0.001
Table 3: Exposures associated with infection, by month (or half-month) of exposure.
Additional information
Closer investigation in the chemotherapy procedures revealed that the outbreak cases were administered different regimens and the chemotherapy preparation was under strict control. This is therefore unlikely that a contaminated batch of chemotherapy drugs could be the source of this outbreak. The cases received chemotherapy on different days, with some overlaps. On the overlapping days often they were treated in different rooms. There was a common preparation area, where auxiliary drugs were prepared.
Minor procedures (e.g. i.v. line flushing, injections) were not fully documented. In e.g. the information was available on placement of an i.v. line, but not about the line care. Multi-dose vials of saline in 100ml containers were used to flush the ports. No syringe re-use was reported.
Actions taken by the hospital
In response to the outbreak the hospital undertook actions to enhance universal precautions with their staff. They also implemented anti-HCV screening at admittance to identify potential chronic infections. Moreover, the multi-dose vials were replaced by single dose vials in this ward.
Discussion
We identified an HCV outbreak in hospital settings confirmed by close genetic relation of the viral strains isolated from the cases. The analysis of exposures points to association of the outbreak with reception of chemotherapy, although the minor procedures performed in relation to chemotherapy administration could be the most likely mechanism of transmission. Two periods are independently associated with infection risk. This implies that the outbreak consisted of at least two transmission events.
Existence of two or more transmission events is also supported by comparing the onset time in symptomatic cases to the diagnosis time in the first diagnosed patient (Pt_05). While an average incubation time for the symptomatic cases implies that transmission could occur in October/November, the first patient was already diagnosed before that time. The likely scenarios include several introductions from the same source-patient or sequential transmission from one patient to another. None of the tested individuals was diagnosed with HCV before. Among the outbreak cases only one had seroconverted by the time of sample collection and this case presented with clinical picture of acute hepatitis C. In the immunocompromised patients seroconversion may be delayed despite ongoing infection [22,26]. However, that should not affect a chronic infection established prior to the hematologic disorder or oncologic treatment. We therefore conclude that the outbreak cases were all acute infections and we did not identify the presumed source patient with chronic infection. This could be expected given that a substantial proportion of the possibly exposed population was not available for screening.
The association of the outbreak strain infection with the number of days of the hospital stay could suggests a repeated or even systematic problem in applying the universal precautions. Neither the hospital infection team nor the outbreak team convened by the local State Sanitary Inspection office was able to identify such procedure. Given the limited number of cases we were not able to study the independent effects of being hospitalized on each particular day, thus the association with the longer hospital stays may also indicate the likelihood of being present on the particular day or days when the transmission event occurred.
Exposure analysis revealed that receiving chemotherapy, especially during one of the implicated time periods was the only factor associated with the risk of infection, that could not be ruled out as contributing to the outbreak. Even if the preparation of chemotherapy was under strict control and fully documented, the minor procedures associated with chemotherapy administration were not. As the cases were administered the chemotherapy in different rooms the supposed breach could occur in medication preparation area, where the only risk factor identified were the multi-dose vials. The link with minor procedures remains hypothetical. However, given the close genetic relation of the HCV strains isolated from the patients and exclusion of other infection mechanisms strongly indicates this transmission route. Moreover, no more cases were identified after enhancement of universal precautions and discontinuation of multi-dose vial use.
Multiple viral outbreaks among patients of oncology and hematology wards have been reported in the literature. The higher prevalence of blood-borne pathogens among patients of such wards may be a contributing factor. HCV infections generally tend to be more prevalent in patients with hematologic malignant diseases as there exists the causative link between HCV and development of certain types of non-Hodgins lymphomas [27]. The transmission routes of the outbreaks quite often are difficult to establish [28–31], but if the mechanisms are clarified, they are most often related to minor procedures, such as syringe reuse or contaminated multi-dose vials, especially used for flushing of catheters or i.v. lines [32–34]. In general, breaches in minor procedures have been implicated in multiple health care related outbreaks [5,30,32,35–37], often linked to sharing vials for several patients or inappropriate environment and hand hygiene. In our report contaminated multi-dose vials remain the most plausible transmission route. Accidental contamination from the surface cannot be excluded, even though clean areas for medication preparation were separated. HCV is able to survive on inanimate surfaces for prolonged time [38], but correct use of antiseptics reduces infectivity to undetectable levels [39]. As the HCV outbreak investigations take place after prolonged time from the exposure proving or disproving surface contamination as source of outbreak was not feasible. Nonetheless, strict adherence to universal precautions and workflow organisation that minimizes the risk of human error should be implemented. Multi-dose vials should be discouraged, especially in the settings where there is higher prevalence of infection among the patient group served, even if safety devices are used.
Finally, we note that the outbreak was identified and investigated with a substantial delay. There were no procedures in place for notification and screening of potentially exposed patients and nearly 40% of the patients could not be reached. Majority of them have died due to their oncologic condition, still analysing their samples and the clinical history would have improved the statistical power to identify the transmission mechanism. Moreover, we may have underestimated the final size of the outbreak, as asymptomatic infections could have occurred in the group that was not screened. Incomplete documentation of the minor invasive procedures constituted another difficulty.
Conclusion
Our results indicate a close relationship of the virus in the haematooncology ward patients suggesting a common source of infection, even though we were not able to fully identify the transmission mechanism. Plausible transmission route includes breaches in minor procedures related to chemotherapy administration and possibly more than one transmission event occurred. As HCV outbreak investigations inevitably rely on medical documentation, we recommend that at least those minor procedures, which were previously linked to transmission, should be documented in detail. Moreover, we note that the investigation was delayed due to difficulties with blood collection from the exposed patients. Procedures on epidemiological management of acute hepatitis C should include sample collection and immediate notification to the local public health departments.
Finally, HCV outbreaks are usually small and dispersed in time, thus molecular analysis, especially with variant analysis is crucial to confirm them.
Acknowledgement
Financial support
K. Caraballo Cortes and M. Radkowski: grants UMO2013/11/B/ NZ6/01679 and UMO-2015/19/D/NZ6/ 01303 from the National Science Center, Poland.
M. Rosinska. M. Stepien. M. Sadkowska-Todys: NIZP-PZH task No. 6/EM.1/2017 and National Health Program task 6/4/3.1k/1/ NPZ/2017/1094/773.
References
- Duffell EF, Hedrich D, Mardh O, Mozalevskis A. Towards elimination of hepatitis B and C in European Union and European Economic Area countries: monitoring the World Health Organization’s global health sector strategy core indicators and scaling up key interventions. Eurosurveillance [Internet]. 2017; 29: 9.
- Wiessing L, Ferri M, Grady B, Kantzanou M, Sperle I, Cullen KJ, et al. Hepatitis C virus infection epidemiology among people who inject drugs in Europe: a systematic review of data for scaling up treatment and prevention. PloS One. 2014; 9: e103345.
- Falla AM, Hofstraat SHI, Duffell E, Hahné SJM, Tavoschi L, Veldhuijzen IK. Hepatitis B/C in the countries of the EU/EEA: a systematic review of the prevalence among at-risk groups. BMC Infect Dis [Internet]. 2018; 291: 18.
- Cornberg M, Razavi HA, Alberti A, Bernasconi E, Buti M, Cooper C, et al. A systematic review of hepatitis C virus epidemiology in Europe, Canada and Israel. Liver Int Off J Int Assoc Study Liver. 2011; 31: 30–60.
- Pozzetto B. Health care-associated hepatitis C virus infection. World J Gastroenterol. 2014; 20: 17265.
- Thompson ND. Nonhospital Health Care–Associated Hepatitis B and C Virus Transmission: United States, 1998–2008. Ann Intern Med. 2009; 150: 33.
- Tavoschi L, Mason L, Petriti U, Bunge E, Veldhuijzen I, Duffell E. Hepatitis B and C among healthcare workers and patient groups at increased risk of iatrogenic transmission in the European Union/European Economic Area. J Hosp Infect [Internet]. 2019.
- Rosinska M, Parda N, Kolakowska A, Godzik P, Zakrzewska K, Madalinski K, et al. Factors associated with hepatitis C prevalence differ by the stage of liver fibrosis: A cross-sectional study in the general population in Poland, 2012- 2016. Khudyakov YE, editor. PLOS ONE. 2017; 20; e0185055.
- Sakem B, Madalinski K, Nydegger U, Stepien M, Godzik P, Kolakowska A, et al. Hepatitis C virus epidemiology and prevention in Polish and Swiss population – similar and contrasting experiences. Ann Agric Environ Med. 2016; 15: 425–431.
- Flisiak R, Halota W, Horban A, Juszczyk J, Pawlowska M, Simon K. Prevalence and risk factors of HCV infection in Poland. Eur J Gastroenterol Hepatol. 2011; 23: 1213–1217.
- Stepien M, Rosinska M. Hepatitis C oubreaks in Poland in 2003-2013. Medical procedures as a dominant route of HCV transmission. Przegl Epidemiol. 2015; 69: 465–472.
- Czerwinski M, Grabarczyk P, Stepien M, Kubicka-Russel D, Tkaczuk K, Brojer E, et al. What weighs more-low compliance with self-deferral or minor medical procedures? Explaining the high rate of hepatitis C virus window period donations in Poland: HCV WINDOW-PERIOD DONATIONS IN POLAND. Transfusion (Paris). 2017; 57: 1998–2006.
- Zakrzewska K, Stepien M, Szmulik K, Rosinska M. Hepatitis C in Poland in 2016. Przegl Epidemiol. 2018; 72: 157–167.
- Stoitsova S, Henszel L, Zakrzewska K, Rosinska M, Duffell E. Healthcareassociated hepatitis B and C transmission to patients in EU/EEA: A systematic review highlights higher-risk settings and variability among countries. In Stockholm, Sweden. 2017; 74.
- Farci P. New Insights into the HCV Quasi species and Compartmentalization. Semin Liver Dis. 2011; 31: 356–374.
- Campo DS, Xia GL, Dimitrova Z, Lin Y, Forbi JC, Ganova-Raeva L, et al. Accurate Genetic Detection of Hepatitis C Virus Transmissions in Outbreak Settings. J Infect Dis. 2016; 213: 957–965.
- Glebova O, Knyazev S, Melnyk A, Artyomenko A, Khudyakov Y, Zelikovsky A, et al. Inference of genetic relatedness between viral quasispecies from sequencing data. BMC Genomics [Internet]. 2017; 6: 18.
- Mahale P, Kontoyiannis DP, Chemaly RF, Jiang Y, Hwang JP, Davila M, et al. Acute exacerbation and reactivation of chronic hepatitis C virus infection in cancer patients. J Hepatol. 2012; 57: 1177–1185.
- Torres HA, Hosry J, Mahale P, Economides MP, Jiang Y, Lok AS. Hepatitis C virus reactivation in patients receiving cancer treatment: A prospective observational study. Hepatol Baltim Md. 2018; 67: 36–47.
- Hoofnagle JH. Course and outcome of hepatitis C. Hepatol Baltim Md. 2002; 36: S21-29.
- Helaly GF, Elsheredy AG, El Basset Mousa AA, Ahmed HKF, Oluyemi AEGS. Seronegative and occult hepatitis C virus infections in patients with hematological disorders. Arch Virol. 2017; 162: 63–69.
- Kazmierczak J, Pawelczyk A, Cortes KC, Radkowski M. Seronegative hepatitis C virus infection. Arch Immunol Ther Exp (Warsz). 2014; 62: 145– 151.
- Caraballo Cortes K, Rosinska M, Janiak M, Stepien M, Zagordi O, Perlejewski K, et al. Next-generation sequencing analysis of a cluster of hepatitis C virus infections in a haematology and oncology center. Khudyakov YE, editor. PLOS ONE. 2018; 13: e0194816.
- Zagordi O, Bhattacharya A, Eriksson N, Beerenwinkel N. ShoRAH: estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinformatics. 2011; 12: 119.
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011; 28: 2731–2739.
- Rossi G, Tucci A, Cariani E, Ravaggi A, Rossini A, Radaeli E. Outbreak of hepatitis C virus infection in patients with hematologic disorders treated with intravenous immunoglobulins: different prognosis according to the immune status. Blood. 1997; 90: 1309–1314.
- Pozzato G. Hepatitis C virus and non-Hodgkin’s lymphomas: Meta-analysis of epidemiology data and therapy options. World J Hepatol. 2016; 8: 107.
- Allander T, Gruber A, Naghavi M, Beyene A, Söderström T, Björkholm M, et al. Frequent patient-to-patient transmission of hepatitis C virus in a haematology ward. Lancet Lond Engl. 1995; 345: 603–607.
- Silini E, Locasciulli A, Santoleri L, Gargantini L, Pinzello G, Montillo M, et al. Hepatitis C virus infection in a hematology ward: evidence for nosocomial transmission and impact on hematologic disease outcome. Haematologica. 2002; 87: 1200–1208.
- Dumpis U, Kovalova Z, Jansons J, Cupane L, Sominskaya I, Michailova M, et al. An outbreak of HBV and HCV infection in a paediatric oncology ward: epidemiological investigations and prevention of further spread. J Med Virol. 2003; 69: 331–338.
- Kološová A, Gašparovič J. Viral hepatitis B and C outbreak related to parenteral treatment at an oncological department in Slovakia. J Hosp Infect. 2016; 93: 211–214.
- Widell A, Christensson B, Wiebe T, Schalén C, Hansson HB, Allander T, et al. Epidemiologic and molecular investigation of outbreaks of hepatitis C virus infection on a pediatric oncology service. Ann Intern Med. 1999; 130: 130–134.
- Macedo de Oliveira A, White KL, Leschinsky DP, Beecham BD, Vogt TM, Moolenaar RL, et al. An outbreak of hepatitis C virus infections among outpatients at a hematology/oncology clinic. Ann Intern Med. 2005; 142: 898–902.
- Rodríguez-Caravaca G, Villar-del-Campo MC, Casas-Losada ML, Cava- Valenciano F, Gil-de-Miguel A. [Hepatitis C outbreak in an oncology ward]. Enferm Infecc Microbiol Clin. 2010; 28: 233–235.
- Centers for Disease Control and Prevention. Healthcare-Associated Hepatitis B and C Outbreaks (= 2 cases) reported to the Centers for Disease Control and Prevention (CDC) 2008-2017 [Internet]. 2018.
- Senatore S, Galli C, Conti A, Faccini M, Cantoni S, Ciconali G, et al. Hepatitis C virus outbreak in a haemodialysis unit: learning from failures. J Hosp Infect. 2016; 94: 249–252.
- Nguyen DB, Gutowski J, Ghiselli M, Cheng T, Bel Hamdounia S, Suryaprasad A, et al. A Large Outbreak of Hepatitis C Virus Infections in a Hemodialysis Clinic. Infect Control Hosp Epidemiol. 2016; 37: 125–133.
- Paintsil E, Binka M, Patel A, Lindenbach BD, Heimer R. Hepatitis C virus maintains infectivity for weeks after drying on inanimate surfaces at room temperature: implications for risks of transmission. J Infect Dis. 2014; 209: 1205–1211.
- Ciesek S, Friesland M, Steinmann J, Becker B, Wedemeyer H, Manns MP, et al. How stable is the Hepatitis C Virus (HCV)? Environmental stability of HCV and its susceptibility to chemical biocides. J Infect Dis. 2010; 201: 1859–1866.
- Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci. 2004; 101: 11030– 11035.