
Case Report
Austin Pediatr Oncol. 2025; 2(1): 1004.
Unilateral Nasal Obstruction in Newborn Revealing an Intranasal Glioma: A Case Report
El Hafi Z*, Nafaa H, Arkoubi Z, Bencheikh R, Benbouzid MA and Essakalli L
ENT doctor in ENT-HNS Department of Specialties Hospital – CHU Ibn Sina Rabat, Morocco, Mohammed V University in Rabat, Morocco
*Corresponding author: Zakaria El Hafi, ENT doctor in ENT-HNS Department of Specialties Hospital – CHU Ibn Sina Rabat, Morocco, Mohammed V University in Rabat, Morocco Email: zakielhafi@gmail.com
Received: March 01, 2025; Accepted: March 24, 2025; Published: March 27, 2025;
Abstract
Heterotopic nasal gliomas are rare congenital malformations presenting as nasal masses composed of heterotopic neuroglial tissue. They are categorized under the nosological framework of midline dysraphisms. These lesions are non-hereditary and are part of a group of malformations affecting the nose and the anterior skull base. This group includes other entities such as dermoïd cysts, encephaloceles, and hemangiomas.
We present the case of a 1-month-old infant that presented to the ENT outpatient consultation for right unilateral nasal obstruction since birth, with difficulty in breathing during breast feeding, aspect upon nasal endoscopy and sino-nasal imaging (CT and MRI) hinted the diagnosis of an intranasal glioma which was confirmed after surgery on histopathological and immunohistochemical reports that showed evidence of glial heterotopia.
The post-operative period was uneventful, and the 6 month follow up nasal endoscopy was normal and showed no signs recurrence
Keywords: Nasal glial heterotopia; Intra nasal glioma; Infant; Endoscopic surgery
Introduction
Neuro-glial heterotopias, also known as nasal glial heterotopias (NGH) or nasal gliomas, are rare congenital, non-neoplastic displacements of cerebral glial tissue in extra cranial sites. The incidence of congenital nasal masses is estimated to be 1 in 20,000 to 40,000 live births, with nasal glial heterotopia (NGH) accounting for approximately 5% of these cases [1]. The nose and nasopharynx are the most commonly involved areas, leading to the frequent use of the term nasal glioma. Other affected sites include the ear, face, neck, and orbit. Due to the rarity of this condition, its diagnosis can often be delayed. This article presents a case report and a literature review of NGH its appropriate diagnostic tools and treatment options, aiming to consolidate current knowledge and provide clearer guidelines for diagnosis and management in infants.
Case Presentation
We present the case of a 1-month-old infant that presented to the ENT outpatient consultation for right unilateral nasal obstruction since birth, with difficulty in breathing during breast feeding, no spontaneous clear rhinorrhea or nose bleeding and no root of nose swelling neurological or ophthalmological signs noted. Physical examination including nasal endoscopy as seen in (Figure 1), revealed a white doughy polyp-like mass occupying the right nasal cavity, anterior to the head of inferior turbinate and with a cranial pedicle that couldn’t be identified clearly during endoscopy. Initially nasal endoscopy didn’t show septal deviation and facial examination showed no deformation of nasal pyramid. Patient was initially treated with saline wash of nasal cavity since steroid spraying isn’t approved under 12 months.
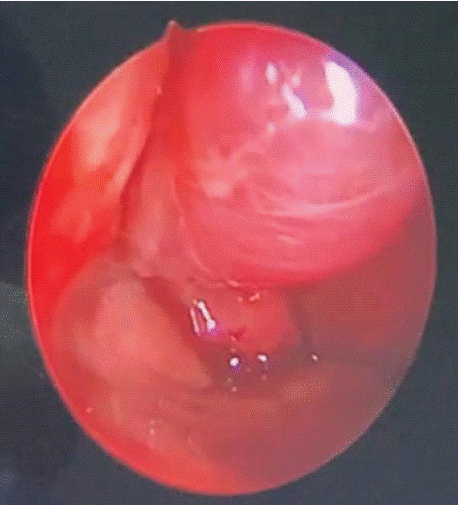
Figure 1: Endoscopic view showing nasal glioma of the anterior part of right
nasal fossa.
Follow up showed no improvement, relatively worsening symptoms and right para nasal deformity appeared between root and nose tip, as shown in clinical photos in (Figure 2).
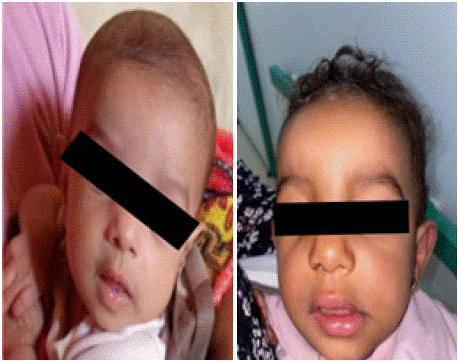
Figure 2: Clinical photos.
Sino nasal computed Tomography and MRI as shown in (Figures 3 & 4); were performed revealing a soft tissue mass in the right nasal cavity in contact with the inferior nasal turbinate seemingly rooted at the roof of the nose. An intracranial connection as well as an encephalocele or meningo-encephalocele were ruled out. No bone abnormality was seen in nasal cavity, base of the skull or paranasal sinuses. The diagnosis of intra nasal glioma was suspected upon history, clinical and imaging findings.
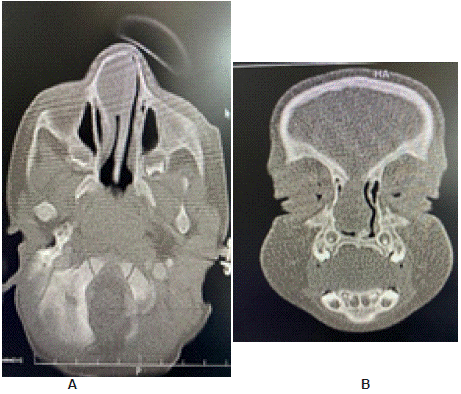
Figure 3: (A) Axial and (B) coronal sections of Naso-cranial CT showing
soft tissue mass in the anterior right Nasal fossa.
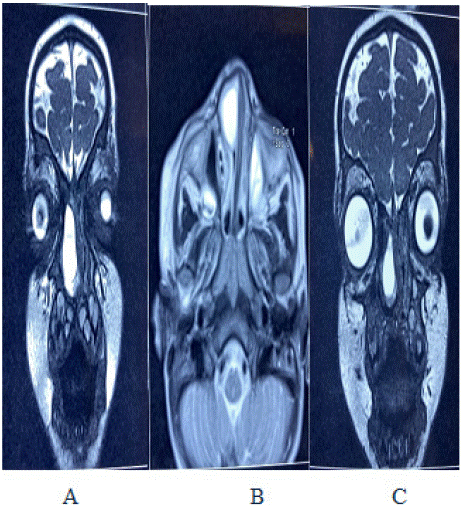
Figure 4: MRI images from left to right showing, A: Coronal section of T2
weighted sequence showing intense soft tissue mass of anterior right nasal
fossa; B: Axial T2 weighted MRI image of the same mass;
C: T2 weighted coronal section showing intense signal soft tissue mass of
the most anterior part of right nasal fossa.
We opted against performing a biopsy and waited for the patient to reach 12 months for technical and anatomical reasons, in order to perform surgery through an exclusive endoscopic approach. The mass was completely resected with clear margins, it’s base was located at the anterior part of the nasal roof, there was no anterior base of skull breach or CSF leak, post-operative follow-up was uneventful, and patient was discharged after 2 days.
Histopathology report analysis confirmed the diagnosis of glial heterotopia, Immunohistochemical staining for glial fibrillary acidic protein (GFAP) confirmed the presence of glial tissue, S100 and synaptophysin and chromogranin A were positive in neurons. There were no signs of malignancy.
6 month follow up endoscopy shows clear right nasal cavity and no sign of recurrence to date.
Discussion
Nasal gliomas were likely first described by Reid in 1852, and the term was officially introduced by Schmidt in 19002,3. Embryologically, the development of nasal cerebral heterotopias is similar to that of nasal encephaloceles or dermoid cysts. During the retraction of the embryonic dural diverticulum, remnants of neural glial tissue become sequestered as their connections to the subarachnoid space are severed and obliterated [1].
The absence of subarachnoid communication distinguishes NGH from anterior encephaloceles.
However, histopathological findings cannot differentiate between gliomas and encephaloceles, as glial tissue may be the predominant or sole component in both lesions [1]. The most common sites involved in nasal gliomas are in and around the nose and nasopharynx, with 60% being extra nasal, 30% intranasal, and 10% involving both regions [2]. Rarely, heterotopic glial tissue can be found in the lips, tongue, scalp, and oropharynx. The incidence of nasal gliomas is one in 20,000 to 40,000 live births, with a higher prevalence in females [3]. Other midline masses that must be considered in the differential diagnosis include hemangiomas, dermoid cysts, and encephaloceles. Therefore, radiological and histopathological examinations are essential for an accurate diagnosis.
Diagnosis of this rare entity relies on nasal endoscopy, MRI, CT and histopathology. Nasal gliomas, despite their name, have no malignant component and do not degenerate. They are mostly asymptomatic but can become infected [4].
The clinical presentation of intranasal gliomas is usually unilateral nasal obstruction as it was the case for our patient. However, over time, these lesions can push the nasal septum and cause bilateral nasal obstruction. Difficulty during breast feeding may also be observed like it was the case for our patient. Occasionally, epistaxis or CSF rhinorrhea may be observed. Recurrent meningitis can at times be the only warning sign. Some authors have suggested a particular tendency for sinusitis in patients with nasal heterotopias [5]. Upon endonasal examination, the mass may be mistaken for a polyp. Gliomas are most often attached to the middle turbinate, but can sometimes develop in contact with the cribriform plate or the nasal septum. In cases of a large gliomas, the nasal bones may be displaced and space between them enlarged. Although the nose is the most common location, other ENT locations such as the nasopharynx, tongue, oropharynx, or scalp have also been described [6].
Extra nasal glial heterotopias are firm, smooth masses that do not pulsate or enlarge during crying, coughing, or straining. They can be associated with hypertelorism [7].
The primary differential diagnosis of heterotopia is nasal hemangioma. Clinically, it poses challenges, and the literature reports numerous diagnostic errors. Non-invasive tests such as Doppler or ultrasound can be useful because hemangiomas exhibit high-velocity arterial flow in late diastole, whereas heterotopia shows low-velocity flow. This distinction is important because while hemangiomas typically regress spontaneously into fibro-fatty tissue, nasal heterotopia does not exhibit any regressive characteristics.
NGHs should be distinguished from dermoid cysts and encephaloceles, as they can all present as midline nasal masses. Encephaloceles are extracranial hernias of the meninges and/or brain due to congenital skull defects. Because they have an intracranial connection, encephaloceles exhibit pulsation and expansion with crying, straining, or compression of the jugular vein (Furstenberg test). It was not the case in our patient as the mass did not pulsate or expand during crying or straining, and there were no developmental anomalies or intracranial connections on NCCT [7].
Nasal dermoid cysts are another differential diagnosis for NGH. Histopathology can differentiate the two: dermoid cysts are lined by keratinized stratified squamous epithelium and contain skin tissues or dermal appendages (e.g., hair follicles, sebaceous glands, and sweat glands), whereas NGH shows mature glial tissue. Rare components reported in NGH include retinal pigmented epithelium, choroid plexus, and ependymal clefts [7].
MRI is superior to CT for acquiring detailed information about soft tissue; it is also more useful for identification of an intracranial connection. On MRI, the lesion shows signal intensity comparable to the brain on T1-weighted images (T1-WI) and high signal intensity on T2-weighted images (T2- WI), featuring many cystic areas and no enhancement. Diffusion-weighted imaging (DWI) reveals no diffusion restriction. MRI is crucial for excluding intracranial communication. Preoperative computed tomography (CT) displays adjacent bony structures and bone defects. On CT images, the lesion appears as a large, well-defined, hypo dense soft-tissue mass. Thus, an MRI is essential, and a CT scan is highly recommended if bony defects are suspected or for navigation support during endoscopic surgery [1].
Previous reviews advise against biopsies of congenital midline masses due to the risk of cerebrospinal fluid leakage if there is an intracranial connection. According to M.G Compte & Al, a biopsy may be recommended, particularly in adults, to exclude malignancy, but should only be performed after appropriate diagnostic imaging (MRI) to rule out an intracranial connection. Early surgical excision is the treatment of choice in nasal glial heterotopias as delay may lead to deformity amongst other complications. For the extra nasal type, if there is no indication of intracranial communication, external or trans facial approaches are sufficient. However, neurosurgical consultation may still be required if an unrecognized tract to the skull base is discovered during surgery. Owing to advancements in endoscopic technology and equipment, intranasal glial heterotopias, as well as mixed NGH can now be adequately visualized and entirely excised [8]. Thus, for small intranasal gliomas without intracranial connections, similar to that of our case, endoscopic excision is advised2.
Follow-up after surgical resection is crucial, as recurrence rates for nasal glial heterotopia range from 4% to 10%. If recurrence is detected during postoperative follow-up, prompt surgical resection should be performed again, and the patient should continue to be closely monitored [9].
Conclusion
Nasal glial heterotopia (NGH) is a rare congenital anomaly that requires a multidisciplinary approach for effective management. Almost 90% of cases are diagnosed before the age of three. Early and accurate diagnosis using MRI is essential to exclude intracranial communication and plan the surgical approach, thereby preventing complications such as cerebrospinal fluid leakage and meningitis. If bony involvement is suspected a CT scan should also be performed.
Surgical resection, using the endoscopic approach, is the definitive treatment for NGH. Complete resection is crucial to minimize the risk of recurrence, and histological evaluation of the surgical margins can further reduce this risk. A follow-up period of one year is typically sufficient, as most recurrences occur within the first 12 months and are usually due to incomplete resection.
Despite its rarity, NGH should always be considered in the differential diagnosis of nasal masses in infants. Prompt diagnosis and surgical intervention are essential to prevent deformities and other complications, ensuring better outcomes for affected infants.
Conception and Design
Zakaria ElHafi, Hiba Nafaa.
Informed Consent
This case report and accompanying images, a copy of the written consent is available for review by the editor-in-chief of this journal on request.
References
- Gallego Compte M, Menter T, Guertler N, Negoias S. Nasal glial heterotopia: a systematic review of the literature and case report. Acta Otorhinolaryngol Ital. 2022; 42: 317‑324.
- Julie CP, Sophie B, Frédérique D, Arnaud G. Nasal glial heterotopia: Four case reports with a review of literature. Oral Maxillofac Surg Cases. 2019; 5: 100107.
- Thaker B, Bhardwaj S. Infantile Nasal Glial Heterotopia - A Case Report. JK Sci J Med Educ Res. 2023; 25: 176‑177.
- Charrier JB, Racy E, Nowak C, Lemaire B, Bobin S. Embryologie et anomalies congénitales du nez. EMC - Oto-Rhino-Laryngol. 2007; 2: 1‑17.
- James Zinreich S, Borders JC, Eisele DW, Mattox DE, Long DM, Kennedy DW. The Utility of Magnetic Resonance Imaging in the Diagnosis of Intranasal Meningoencephaloceles. Arch Otolaryngol - Head Neck Surg. 1992; 118: 1253‑1256.
- Kennard CD, Rasmussen JE. Congenital Midline Nasal Masses: Diagnosis and Management. J Dermatol Surg Oncol. 1990; 16: 1025‑1036.
- Tahlan K, Tanveer N, Kumar H, Diwan H. A Rare Case of Nasal Glial Heterotopia in an Infant. J Cutan Aesthetic Surg. 2020; 13: 233.
- Intranasal glial heterotopia in an infant boy [Internet]. 2024.
- Yan Y yan, Zhou Z ying, Bi J, Fu Y. Nasal glial heterotopia in children: Two case reports and literature review. Int J Pediatr Otorhinolaryngol. 2020; 129: 109728.