
Review Article
J Hepat Res. 2024; 8(1): 1052.
Hepatotoxicity Associated with First-Line Anti-Tuberculosis Drugs and the Role of Hepatitis B and C Virus Infections: A Review
Birhanu Getie¹*; Etenesh Wondimu²; Andargachew Almaw¹; Mulat Erkihun¹; Biruk Legesse¹; Ayenew Berhan¹
¹Department of Medical Laboratory Science, College of Health Science, Debre Tabor University, Debre Tabor, Ethiopia
²College of Veterinary Medicine, Bonga University, Bonga, Ethiopia
*Corresponding author: Birhanu Getie, Department of Medical Laboratory Science, College of Health Science, Debre Tabor University, Debre Tabor, Ethiopia. Email: bireget95@gmail.com
Received: October 14, 2024; Accepted: November 01, 2024 Published: November 08, 2024
Abstract
The liver’s vital function in drug metabolism and detoxification makes it vulnerable to injury. Drug-induced liver damage can be caused by the primary drug’s direct toxicity, immune-mediated responses, or metabolites. One typical side effect of first-line anti-TB drugs is hepatotoxicity. The current first-line medications for treating tuberculosis are isoniazid, pyrazinamide, ethambutol, streptomycin, and rifampicin. Cytochrome P-450 is the most important family of liver enzymes involved in the metabolism of anti-tuberculosis medications. HBV carriers are more susceptible to hepatotoxicity from first-line anti-tuberculosis drugs due to the possibility of HBV reactivation, increased immune system function from TB infection control, decreased metabolism of first-line anti-TB drugs, and pro-inflammatory conditions caused by HBV replication. Symptoms of liver damage often include nausea, vomiting, jaundice, and pain in the abdomen. Liver-protecting drugs such as silymarin and N-acetyl cysteine may be able to reduce the damage that first-line anti-TB drugs cause to the liver by removing the toxin from the liver and improving the regeneration of liver cell membranes. The earliest possible cessation of the offending medicine is the most crucial step in addressing hepatotoxicity. This review tries to compile and provide the aggregate results of multiple literatures from different regions of the world in order to give baseline information. As a result, it is based on relevant literature that was found using the following keywords in a separate and combined search: “incidence, pathology, first-line anti-TB drug-induced hepatotoxicity, first-line anti-TB drug metabolism, mechanisms of toxicity, hepatitis B and C virus infections.” These databases included PubMed/Medline, the Cumulative Index to Nursing and Allied Health Literature, African Journals Online, and Google Scholar. References were made using the EndNote referencing manager. Studies show that the prevalence of hepatotoxicity ranges widely between countries, from 2% to 39%. There is a chance that rifampicin, isoniazid, and pyrazinamide will cause liver damage. Hepatotoxicity has not been observed in relation to ethambutol or streptomycin. The hepatitis B and C infections enhance the risk of getting drug-induced hepatotoxicity when using TB treatment. An increase in the patient’s alanine transaminase level is a sign that liver protectors should be used. Routine monitoring of liver function and hepatitis virus load is recommended during tuberculosis treatment.
Keywords: Anti-Tuberculosis drugs; Drug-induced hepatotoxicity; Hepatitis B and C virus
ABBREVIATIONS: ADR: Adverse Drug Reaction; ALP: Alkaline Phosphatase; ALT: Alanine Transaminase; AST: Aspartate Transaminase; CHB: Chronic Hepatitis B; CLD:Chronic Liver Disease; DIH: Drug-Induced Hepatotoxicity; DILI: Drug Induced Liver Injury; DNA: Deoxyribonucleic Acid; EMB: Ethambutol; FATDH: First Line Anti- Tuberculosis Drug Induced Hepatotoxicity; HBsAg: Hepatitis B Surface Antigen; HBV: Hepatitis B Virus; HCC: Hepatocellular Carcinoma; HCV: Hepatitis C Virus; HIV: Human Immunodeficiency Virus; INH: Isoniazid; LTBI: Latent Mycobacterium Tuberculosis Infections; PZA: Pyrazinamide; RMP: Rifampicin; RNA: Ribonucleic Acid; sGOT: Serum Glutamic Oxaloacetic Transaminase; sGPT: Serum Glutamate Pyruvate Transaminase; ssRNA: Single Stranded Ribonucleic Acid; TB: Tuberculosis; TLI: Transient Liver Function Impairment; ULN: Upper Limit of Normal range; WHO: World Health Organization.
Introduction
Mycobacterium Tuberculosis (Mtb) is the infectious agent that causes Tuberculosis (TB). In both industrialized and developing nations, it continues to be a major health issue [1]. A projected 10.6 million people contracted tuberculosis in 2021 as opposed to 10.1 million in 2020, and 10.5 million people died from tuberculosis in 2020 as opposed to 10.5 million in 2021 (including 214 000 HIVpositive individuals). Furthermore, compared to 2020, the incidence rate of tuberculosis increased by 3·6% in 2021, indicating a reversal from the pattern of roughly 2% annual decline over the previous two decades [2]. Primary anti-tuberculosis medications possess potent bactericidal properties. Currently, Isoniazid (INH), Rifampicin (RMP), Pyrazinamide (PZA), and Ethambutol (EMB) are advised as first-line treatments for Tuberculosis (TB) for a period of two months, followed by a four-month period of INH, RMP, and/or EMB [3].
Hepatotoxicity, derived from hepatic toxicity, denotes liver damage caused by chemicals. Both acute and chronic liver disease can be brought on by drugs that cause liver damage. An elevated Alanine Transaminase (ALT) or Aspartate Transaminase (AST) of three times the Upper Limit of Normal range (ULN) with symptoms (e.g., nausea, vomiting, abdominal pain, unexplained fatigue, or jaundice) related to liver injury or five times the ULN of ALT or AST without symptoms has been used in the majority of reports to define hepatotoxicity [4]. Increases in aspartate Aminotransferase (AST) or Serum Glutamic Oxaloacetic Transaminase (sGOT), which can also indicate problems in the heart, kidney, or muscle, are less specific for hepatocellular injury than increases in serum ALT, formerly known as Serum Glutamate Pyruvate Transaminase (sGPT) [5].
One of the most common and dangerous side effects of first-line anti-TB medications is hepatotoxicity, which can cause abrupt liver failure. It can also compromise treatment regimens and possibly lead to drug resistance, which can diminish the effectiveness of treatment. Withdrawing from anti-TB medication increases the risk of developing multidrug-resistant tuberculosis, which can occur when anti-TB treatment-induced hepatitis symptoms appear. The liver is mostly responsible for this drug's metabolism. Drugs or drug metabolites are extremely harmful to hepatocytes because the enzymes involved in drug metabolism in hepatocyte microsomes may be congenital, malformed, have poor activity, or be blocked by drugs [6].
Liver is prone to damage since it plays a crucial part in drug metabolism and detoxification. Hepatic adaptability and hepatocellular damage are two different drug-induced liver injury pathologies. It is essential to understand the processes and metabolism of anti-TB drugs in DILI [7]. There have been conflicting reports about the frequency of DIH (first-line anti-TB drug-induced hepatotoxicity) during regular multimodal TB treatment [8]. Patients with hepatotoxicity risk factors experience hepatotoxicity from first-line anti-TB drugs much more frequently and with greater severity. The following were listed as risk factors for the development of DILI during first-line anti- TB treatment: extra-pulmonary TB, slow acetylators status, increased serum transaminases before treatment, advanced age, female gender, alcohol misuse, malnutrition, and HIV infection [9,10].
Infections with the hepatitis B and C viruses can raise the risk of hepatotoxicity. When receiving first-line anti-TB medication, HCV infection significantly increases the likelihood of having either transitory liver impairment or DILI. The enveloped +ssRNA Hepatitis C virus (HCV) belongs to the Flaviviridae family and exhibits a high level of genetic variability [11]. It results in cirrhosis, hepatocellular cancer, and chronic hepatitis [12]. The Hepadnaviridae family of viruses includes the enveloped DNA virus known as the Hepatitis B Virus (HBV). The Hepatitis B Virus (HBV) causes persistent infections and has a significant role in the eventual development of hepatocellular carcinoma and liver damage [13]. During first-line anti-TB treatment, the hepatitis B virus (HBV) increases the risk of developing abnormal Liver Function Tests (LFTs) and mortality [14]. Individuals who co-infect HBV and have chronic HBV are more likely to get liver failure and poor treatment results while starting first-line anti-TB medication [15].
The primary limitations of this review are the lack of information about the hepatotoxicity of second-line anti-TB medications and the disarray of data about each component. Nevertheless, the goal of this review is to provide a baseline of knowledge on the subject by summarizing and presenting the collective findings of multiple international literatures. This review is crucial because it gives clinicians thorough information about the issues TB patients confront during treatment, including hepatotoxicity linked to viral hepatitis and problems with anti-TB drugs. Furthermore, it might be crucial in lowering the mortality rate of TB patients.
The aim of this review is to assess the hepatotoxicity of firstline anti-TB medications and the impact of hepatitis B and C virus infections. Electronic databases such as PubMed/Medline, African Journals Online, Cumulative Index to Nursing and Allied Health Literature, and Google Scholar were searched in order to compile the relevant literature on the subject of this review using the key terms "incidence, pathology, clinical features, first line anti-TB drug induced hepatotoxicity, first line anti-TB drug metabolism, mechanisms of toxicity, hepatitis B virus and hepatitis C virus" separately and in combination. The articles were read in their entirety to determine eligibility. English-language articles were consulted in composing this review. Not included were any articles that were not publicly accessible. Apart from complete studies, there are abstracts accessible concerning hepatotoxicity caused by first-line anti-TB drugs and the involvement of HCV or HB. For reference, the EndNote referencing manager was utilized.
Incidence, Pathology and Clinical Features of First Line Ant-Tb Drugs Induced Hepatotoxicity
Incidence of First Line Ant-TB Drugs Induced Hepatotoxicity
Estimating the incidence of hepatotoxicity caused by particular medicines is challenging because most patients receive a combination of medications during their TB therapy. Many medications are often used to treat active tuberculosis. As a result, information regarding the individual toxicity rates of first-line anti-tuberculosis medications is scarce, with the exception of isoniazid. This may make it more difficult to link the reaction to a particular drug. Temporal correlations are the only way to show that a particular medicine is the cause of an unfavorable impact.
The majority of research on First-line Anti-Tuberculosis Drugs Causing Hepatotoxicity (FATDH) has been conducted in China, with varying incidences seen. The greatest rates (71.59%) were likewise seen in research conducted in China. With a cumulative frequency of 2.55%, 106 patients out of 4,304 TB patients getting Directly Observed Treatment Strategy (DOTS) treatment experienced DILI. In the first two months following the start of treatment, 71.59% of patients experienced DILI; the median amount of time between the start of first-line anti-TB treatment and ALT rise was 52.50 days [16]. 7 (8.7%) of the 195 TB patients in the research demonstrated hepatotoxicity, and the mean aspartate aminotransferase/alanine aminotransferase levels in the hepatotoxicity group were 249/249 IU/L, respectively. These findings are from a study conducted in Korea. Twelve of the 17 hepatotoxic patients had first-line anti-TB DIH. Two individuals had hepatotoxicity associated with RMP or INH, while ten patients had hepatotoxicity related to PZA [17].
46.7% of 120 patients in an Indian study reported Adverse Drug Reactions (ADRs) to first-line anti-tuberculosis medications. Three points were used to rank the severity of ADRs: mild (34.2%), moderate (9.5%), and severe (3.3%). Three patients (2.5%) suffered hepatotoxicity. It was discovered more frequently during the intensive phase and was serious enough to require stopping anti-tubercular therapy due to the development of fever, anorexia, jaundice, and impaired mental status [18]. In South Africa, out of 8984 children treated for tuberculosis, 75 (0.83%) had jaundice recorded, and 380 (9.9%) had abnormal liver function tests. The majority of the patients received INH treatment, but there were also different combinations of INH, RMP, and PZA [19].
An observational study conducted in Ethiopia found that 159 patients (15%) had DILI, with severity ratings 1, 2, and 3 accounting for 53.5%, 32.7%, 11.3%, and 2.5% of cases, respectively. Cholestasis, hepatocellular, and mixed pattern incidences were 61%, 15%, and 24%, respectively [20]. According to several studies, hepatotoxicity caused by first-line anti-TB medications is typically seen in the first eight weeks of treatment. In research conducted at Jimma University Hospital, the incidence of hepatotoxicity was 11.5%, and the mean interval between the start of first-line anti-TB medications and the rise of aminotransferases was 26 days. The majority of hepatotoxicity incidences (93.9%) happened during the treatment's intensive phase [21]. It was discovered that the anti-TB-DIH incidence was 8% in the Dawro Zone, South Ethiopia. In addition to hepatotoxicity signs and symptoms (nausea, vomiting, anorexia, malaise, and jaundice), the subjects also had elevated serum transaminase and bilirubin levels. Hepatotoxicity began 13 days to 58 days (median, 26 days) following the start of treatment [22].
ncidence of Hepatotoxicity Attributable to a Specific First- Line Anti -Tuberculosis Drugs
The investigation comprised 195 patients from a tertiary hospital. Nineteen patients, or 9.7% of the total, had hepatotoxicity. The hepatotoxicity group's mean AST and ALT levels were 245 and 244, respectively. Of the 19 patients, 8 had hepatotoxicity linked to pyrazinamide, 9 had hepatotoxicity related to INH or RMP, and 2 did not have an incident connected to anti-TB drugs [23]., One common prophylactic monotherapy for latent tuberculosis infections is isoniazid. The majority of current research has shown that INH monotherapy can cause symptomatic hepatotoxicity at rates between 0.1% and 0.3%, with 10% to 20% of cases showing subclinical elevations in serum transaminases [24]. Patients getting rifampicin along with other anti-TB medications have been found to have a higher incidence of hepatotoxicity, which is thought to be more than 4%. The incidence of symptomatic hepatitis and elevated serum aminotransferase has been observed to range from 0.6% to 2.7% when rifampicin is taken as monotherapy for cholestatic pruritus [25]. 48 occurrences of hepatotoxicity were linked to a 2-month course of rifampicin-pyrazinamide therapy for latent tuberculosis, according to Centers for Disease Control update. 37 patients made a full recovery, while 11 passed away from liver failure. 33 (69%) of the 48 cases that were recorded happened during the second month of treatment. By itself, pyrazinamide is a reasonably safe medication. Pyrazinamide and rifampicin together are linked to increased toxicity (7.7% severe hepatotoxicity) [26]. Fewer cases of hepatotoxicity with ethambutol therapy for tuberculosis have been documented. Some ethambutol users have reported abnormal liver function tests; however, these patients were also prescribed other anti-TB medications that are known to induce liver impairment. There have been no reports of streptomycin hepatotoxicity [25].
Pathological Features of First Line Ant-TB Drugs Induced Hepatotoxicity
Drug-Induced Liver Injury (DILI) affects hepatocytes, biliary epithelial cells, and/or the liver vasculature and is caused by the direct toxicity of the original chemical, a metabolite, or an immunemediated reaction. Reactions that are erratic or unique make up the majority of DILI types. These hypersensitivity or metabolic responses, which cause cholestasis and/or hepatocellular damage, are infrequent and mostly dose-independent for each medication. As is frequently observed with predicted DILI, hepatocyte necrosis is frequently dispersed across the hepatic lobules as opposed to being zonal. Immunogenic drugs or their metabolites covalently bind to liver proteins in hypersensitivity reactions, generating haptens or "neoantigens." It is possible to elicit T-cell, antibody-dependent cytotoxic and occasionally eosinophilic hypersensitivity responses. Interleukin (IL)-12, released tumor necrosis factor, and IFN stimulate hepatocellular programmed cell death (apoptosis), an effect that is countered by IL-4, IL-10, IL-13, and monocyte chemotactic protein-1 [27].
It has been postulated that toxic isoniazid metabolites bind covalently to cell macromolecules. Case studies including both humans and animals demonstrate that isoniazid-induced hepatotoxicity primarily appears as hepatic steatosis and necrosis. The suspected toxic isoniazid metabolite is hydrazine. Hydrazine produces steatosis, hepatocyte vacuolation, glutathione depletion, and inflammatory infiltrates with a high concentration of eosinophil. Periportal and midzonal hepatocytes have lipid vacuoles and swollen mitochondria [28]. Rifampicin may interfere with bilirubin excretion, resulting in temporary hyperbilirubinemia. However, this is not a harmful side effect. Hepatic lesions caused by rifampicin may include centrilobular necrosis, hepatocellular alterations, and perhaps cholestasis. The histopathological results exhibit varying degrees of necrosis, from patchy to diffuse, with nearly total cholestasis. Focal cholestasis, lymphocytic infiltration, bridging necrosis, increased fibrosis, and micro nodular cirrhosis were observed in the liver of a patient who died of rifampicin and pyrazinamide-induced hepatotoxicity [29].
Clinical Features of First Line Ant-TB Drugs Induced Hepatotoxicity
The clinical presentation of First line Anti- Tuberculosis Drug Induced Hepatotoxicity (FATDH) is similar to that of acute viral hepatitis. First line anti TB drug-induced hepatotoxicity can be confirmed if the levels of liver enzymes normalize and the signs and symptoms of hepatotoxicity are resolved after the withdrawal of all first line anti-tubercular drugs. First line anti-tuberculosis treatments can cause varied degree of hepatotoxicity from a transitory asymptomatic rise in transaminases to acute liver failure. Hepatic drug reactions usually occur in the first 2 months of treatment but may happen at any moment during the treatment period. Fatality due to first line anti-TB drugs-induced hepatotoxicity was found to be more likely when jaundice occurred after 6 weeks of the initiation of therapy, serum bilirubin levels were higher, or the treatment was continued despite jaundice. Jaundice, abdominal discomfort, nausea, vomiting, asthenia, lethargy, weakness, right upper quadrant pain, itching, skin rash, anorexia, and weight loss are the telltale signs and symptoms of liver damage. They are insufficiently specific to diagnose a liver condition. As a result, laboratory liver testing is needed for confirmation. Interrupting treatment usually relieves FATDH complaints. FATDH can be lethal if medication is not stopped in a timely manner [30,31].
Overall Metabolism and Mechanisms of Toxicity
Mechanism
For most medications, idiosyncratic reactions and direct hepatotoxicity are part of the pathophysiology of liver injury; however, for certain treatments, the mechanism of harm is inferred based on clinical presentation and hepatic histology results [32].
Because of its distinct metabolism and intimate connection to the gastrointestinal system, medications and other chemicals can harm the liver. Nearly pure forms of medications and xenobiotics are transported to the spleen by portal veins, which carry 75% of the blood that goes to the liver from the gastrointestinal tract. Numerous substances harm the mitochondria, an organelle inside the cell that generates energy. When it malfunctions, an excessive amount of oxidants are released, harming the liver cells. Oxidative stress is also brought on by the activation of several cytochrome P-450 system enzymes, such as CYP2E1. Bile acid buildup in the liver is caused by damage to hepatocyte and bile duct cells. This encourages more liver damage. Non-parenchymal cells with a similar function include Kupffer cells, stellate cells that store fat, and leukocytes (such as neutrophils and monocytes) also have a role in the mechanism [33].
Drug Metabolism in the Liver: Almost all medications are recognized by the human body as alien substances (also known as xenobiotics), and they undergo a number of chemical reactions in order to prepare them for excretion. Chemical changes are necessary in order to: (a) decrease fat solubility; and (b) alter biological activity. The primary "metabolic clearing house" for both endogenous molecules (such as cholesterol, steroid hormones, fatty acids, and proteins) and foreign compounds (such as medications, alcohol) is the smooth endoplasmic reticulum in the liver. The most significant family of metabolizing enzymes in the liver is cytochrome P-450, a collection of enzymes found in the endoplasmic reticulum. It is actually made up of 50 isoforms that are closely linked to one another rather than being a single enzyme. Due to this heterogeneity, the liver can oxidize a wide range of substances in phase 1, including practically all medications. Enzyme inhibitors prevent one or more P-450 enzymes from being metabolized. Isoniazid, for instance. Conversely, inducers boost the production of P-450, which raises its activity [34,35].
Metabolism and Toxicity Mechanism of First-Line Anti-Tb Drugs
The liver is situated between the alimentary tract and the systemic circulation to maximize processing of absorbed nutrients and to minimize exposure of the body to toxins and foreign chemicals. The splanchnic circulation carries ingested drugs directly into the liver. Metabolic enzymes convert these chemicals through phase 1 pathways. The formation of reactive metabolites has been implicated in a range of clinical toxicities. Reactive metabolites are generally electrophile. When they escape detoxification, they react with nucleophilic groups such as lysine and cysteine on cellular proteins. Covalently modified cellular proteins can either be repaired or degraded. If these processes fail, drug-metabolite adduct formation itself impairs important cellular function, leading to the manifestation of target organ injury. Generation of reactive metabolites followed by covalent protein binding can also lead to immune-mediated injury. High levels of reactive metabolite formation in an individual may be due to high levels or increased activities of enzymes involved in the biotransformation of a drug into a reactive metabolite; these are generally phase I cytochrome P450 enzymes involved in oxidation, reduction, or hydrolysis. Alternatively, individuals may have low levels or reduced activities of enzymes that detoxify reactive metabolites, usually mediated by phase II enzymes through a process of glucuronidation, sulfation, acetylation, or glutathione conjugation. Phase III of drug disposition is mediated by transporter molecules or proteins, which facilitate excretion of the water-soluble metabolites into bile or systemic circulation. Most first-line anti-TB drugs are lipophilic, and their biotransformation involves their conversion into water-soluble compounds and subsequent elimination. Hepatotoxicity appears to involve reactive metabolite formation and accumulation rather than the direct effect of the parent drug itself [36,37].
Isoniazid
Metabolism: Isoniazid is cleared mostly by the liver, primarily via acetylation by N-Acetyl Transferase 2 (NAT-2). Acetyl-isoniazid is metabolized mainly to Mono-Acetyl Hydrazine (MAH) and to the nontoxic diacetyl hydrazine. Individuals with prolonged t1/2 have extended exposure to the drug. Genetic polymorphisms of NAT-2 correlate with fast, slow, and intermediate acetylation phenotypes. Microsomal enzymes (e.g., cytochrome P450 2E1) further metabolize isoniazid intermediates through phase 1 pathways. The plasma halflife of AcHz (a metabolite of INH) is shortened by Rifampicin, and AcHz is quickly converted to its active metabolites by increasing the oxidative elimination rate of AcHz, which is related to the higher incidence of liver necrosis caused by INH and Rifampicin in combination. In fast acetylators, more than 90% of the drug is excreted as acetyl-isoniazid, whereas in slow acetylators, 67% of the drug is excreted as acetyl-isoniazid, and a greater percentage of isoniazid is excreted as unchanged drug into the urine. Slow acetylation results not only in accumulation of the parent compound but also of monoacetyl hydrazine. Acetylation of acetyl hydrazine is further suppressed by INH itself. In addition, direct hydrolysis of INH without acetylation produces hydrazine that could cause liver injury. Fast acetylators clear MAH more rapidly. Slow acetylators may actually have greater cumulative MAH exposure. Slow acetylators are a risk factor for DIH, and patients are prone to developing more severe hepatotoxicity than with rapid acetylators [37,38].
Mechanism of injury: Reactive metabolites of MAH are probably toxic to tissues through free radical generation in rats, the free radical scavenger glutathione-related thiols and antioxidant glutathione peroxidase and catalase activities are diminished by isoniazid, although glutathione reductase activity is increased. The antioxidant N-acetyl-cysteine, a substrate for glutathione synthesis, inhibits isoniazid-induced liver injury in pretreated rats, with unknown relevance in humans. The isoniazid metabolite acetyl-hydrazine covalently binds to liver macromolecules, a process mediated by microsomal enzymes. Patients with homozygous cytochrome P450 2E1 host gene polymorphism, who have enhanced cytochrome P450 2E1 activity, in one study had a higher risk of hepatotoxicity, particularly in slow acetylators [38,39].
Rifampicin
Metabolism: Rifampicin is well absorbed from the stomach and metabolized in the liver by desacetylation to desacetyl rifampicin, and a separate pathway of hydrolysis produces 3-formyl rifampicin. Desacetyl rifampicin is more polar than the parent compound and microbiologically active. These metabolites are non-toxic. The xeno sensing pregnane X receptor (PXR) is a member of the nuclear receptor superfamily of ligand-dependent transcription factors that can be activated by a variety of drugs including rifampicin. Activated PXR binds to response elements in the promoters and upregulates the transcription of phase I and II drug metabolizing enzymes such as Cytochrome P450 (CYP)s, Glutathione S-Transferases (GSTs), and transporters (involved in phase III). Rifampicin is a potent inducer of several metabolic enzyme pathways, in particular the Cytochrome P450 (CYP3A4) system via the hepatocyte PXR. This activation of the CYP3A4 leads to increased metabolism of isoniazid yielding toxic metabolites, thus explains the potentiating effect of rifampicin in anti-TB drug- induced hepatotoxicity. Rifampicin also induces isoniazid hydrolases, leading to increased hydrazine production, especially in slow acetylators, thus increasing the toxicity when used in combination with isoniazid. Rifampicin occasionally interferes with bilirubin uptake and results in transient unconjugated hyperbilirubinemia without hepatocyte damage. However, more commonly, it does contribute to conjugated hyperbilirubinemia via interfering with the bilirubin excretion by inhibiting the Bile Salt Exporter Pump (BSEP) [40,41].
Mechanisms of hepatotoxicity: Rifampin is the cause of conjugated hyperbilirubinemia because it inhibits the primary bile salt exporter pump. In addition, dose-dependent competition with bilirubin for clearance at the sinusoidal membrane or impaired secretion at the canalicular level may cause asymptomatic increased bilirubin levels. It seems that rare hepatocellular damage is a hypersensitivity reaction, and that large, sporadic dosages may make it more likely. Hemolytic anemia and renal impairment have been documented in conjunction with hypersensitivity reactions [42].
Pyrazinamide
Metabolism: Pyrazinamide has a half-life (t1/2) of roughly 10 hours, which is noticeably longer than that of isoniazid or rifampin. For individuals who already have hepatic illness, t1/2 is extended to 15 hours. Pyrazinamide is a derivative of nicotinic acid that is deamidated to pyrazinoic acid in the liver. Xanthine oxidase, aldehyde oxidase, and xanthine dehydrogenase then convert pyrazinoic acid to 5-hydroxy-pyrazinoic acid. In addition, 5-hydroxy-pyrazinamide may be produced during metabolism. Pyrazinamide's serum half-life does not correlate with treatment duration, suggesting that the drug does not stimulate the enzymes needed for its metabolism. Pyrazinamide reduced CYP450 activity in mouse models, and changes in NAD levels were linked to hepatotoxicity caused by free radical species. Patients with renal impairment must take pyrazinamide on an intermittent basis because the kidneys remove its metabolites [38,43].
Mechanism of injury: Pyrazinamide may cause hepatotoxicity that is both idiosyncratic and dose dependent. Daily doses of pyrazinamide at 40 to 50 mg/kg were linked to a higher incidence of hepatotoxicity several decades ago than the doses used in current regimens (25–35 mg/kg). In rat liver, pyrazinamide changes the levels of nicotinamide acetyl dehydrogenase, potentially leading to the production of free radical species. Isoniazid may cause harm through comparable pathways due to some chemical structural similarities. Patients treated with rifampin plus pyrazinamide for LTBI have experienced more severe side effects than those who previously experienced hepatotoxic responses with isoniazid. Pyrazinamide may cause granulomatous hepatitis, liver damage, and hypersensitivity reactions accompanied by eosinophilia [43,44].
Prophylactic Treatment with Rifampicin and Pyrazinamide
The standard treatment for latent Mycobacterium tuberculosis infections is isoniazid monotherapy for six months. Investigations on a two-month preventive regimen involving pyrazinamide and rifampicin resulted in fatal and extremely significant cases of hepatotoxicity. In comparison to six months of isoniazid treatment (8–13% versus 1-4%), it resulted in more frequent and severe hepatotoxicity. It even produced more hepatotoxicity when compared to normal treatment for active tuberculosis. The reason why a 6-month regimen containing isoniazid, rifampicin, and pyrazinamide is less harmful than rifampicin and pyrazinamide alone is yet unknown. According to some writers, pyrazinamide might be the main factor. It is also possible to think of a pharmacological interaction in which isoniazid reduces the hepatotoxic potential of pyrazinamide and rifampicin, albeit the exact processes involved are unknown [26,45].
First Line Ant-Tb Drugs Induced Hepatotoxicity and The Role of Hepatitis B and C Viruse Infections
The incidence of first-line anti-TB induce hepatoxicity is higher in developing countries compared to those of developed countries. One possible explanation is the higher prevalence of viral hepatitis in developing countries [46]. Hepatitis B and C viruses are risk factors for the development of abnormal LFTs and mortality during first-line anti-TB treatment [14]. Pulmonary tuberculosis patients with HBV were more sensitive to hepatotoxic drugs because of pre-existing hepatic damage, and the liver function of these patients improved more slowly. Patients on first-line anti-TB therapy with chronic HBV co-infection are more susceptible to developing liver failure and having poor outcomes during TB treatment. TB-HBV group were more susceptible to Grade-4 severity of DILI (36.2% vs. 7.7%), liver failure (67.2% vs.38.5%), and poor outcomes (37.9% vs. 7.7%), compared with patients in the TB group. Patients in the TB-HBV group had a higher rate of death than those in the TB group (34.5% vs. 7.7%). Advanced age, cirrhosis, and severe hyperbilirubinemia were independent risk factors for the incidence of death in the TB-HBV group [15].
In a study in China, the incidence of Transient Liver Function Impairment (TLI) was significantly lower in controls than in chronic hepatitis patients (2% vs. 12%). The mean onset times of DIH in the control HBV, and HCV groups were not significantly different (40, 39, and 67 days, respectively). The mean onset times of TLI in the control, HBV, and HCV groups were significantly different (23, 48, and 68 days, respectively,). Liver function impairment during anti- TB therapy in patients with chronic viral hepatitis was due to mostly TLI, with TLI occurring later than in controls [47]. Another study in China showed 40 HCV-seropositive patients (74%) and 82 control subjects (85%) received an initial treatment regimen that included pyrazinamide. Twenty-two HCV-seropositive patients (41%) and 19 control subjects (20%) exhibited elevated liver enzyme levels during TB treatment, including transient elevation of transaminase [48].
Most of the hepatocytes in HBV carriers without clinical symptoms had changes in histology and spot necrosis. One researcher took liver biopsy from 25 pulmonary tuberculosis patients with HBV infection during the course of first-line anti-TB treatment and discovered that all the patients with liver dysfunction suffered from viral hepatitis, even liver cirrhosis. Hepatic damage in the patients was related to HBV infection and pre-existing pathologic changes in the liver. Firstline anti-TB medicines only aggravated pre-existing hepatic damage. Hepatic damage in the patients with positive HBV was caused by viral damage overlapped by medicine damage. The rate of hepatotoxicity occurred in 26 (59%) TB patients with HBV during first- line anti- TB treatment, higher than that in 40 (24%) TB patients without HBV. Hepatotoxicity caused by first-line anti-TB medicines usually happens in the first 2-3 months of TB treatment. In the study, 66 out of 217 patients had hepatic damage, which happened in 21 patients within 1 month, in 45 patients within 2 months [49,50].
In Taiwan, 42 (2.4%) of 1,783 patients with TB treated with isoniazid, rifampin, and ethambutol had symptomatic hepatitis. Fifteen were hepatitis B carriers (had hepatitis B surface antigen), and 7 of 15 died of hepatic failure. Of the other 27 patients with symptomatic hepatitis who were not hepatitis B carriers, one died of hepatic failure. The severity of hepatotoxicity appears to have been increased in the hepatitis B carrier population [51]. Also, in Taiwan, hepatitis B carriers with TB who received isoniazid, rifampin, pyrazinamide, and ethambutol had a hepatotoxicity rate of 29%, compared to the 26% experienced by hepatitis B-seronegative individuals. Patients were excluded if alcohol ingestion exceeded 60 g/day or if baseline serum transaminase concentrations were greater than the ULN [52].
In a study from Hong Kong, which excluded alcoholic and nonviral liver diseases, 16% of patients with TB with hepatitis B surface antigen developed symptomatic hepatitis compared with 4.7% in those without hepatitis B infection. Patients who had hepatitis B surface antigen also had more severe liver injury and were more likely to have a permanent treatment discontinuation, 4.7 compared with 2.5% [50]. A study from Korea of 110 patients with hepatitis B surface antigen and normal pretreatment transaminases found a trend toward transaminase elevations of at least five times the ULN more frequently in the hepatitis B carrier group than in the control subjects (8 vs. 2%). However, isoniazid and rifampin were successfully reintroduced in five of the nine carriers [53].
The reason why HBV carriers have a higher risk of First-Line Anti-TB-Induced Liver Injury (FATDILI) remains to be elucidated. However, several explanations have been proposed, including a suspicion about reactivation of HBV with flare-up hepatitis, which has manifested with high HBV-DNA and positive HBeAg in some patients as mentioned. However, many HBV carriers with liver dysfunction had negative HBeAg and low or undetectable HBV-DNA, whose hepatic damage could not be attributed to HBV. In addition, the HBV carriers with first-line anti-TB treatment still had a higher incidence of liver injury than the HBV carriers without first-line anti- TB therapy in the same follow-up period. Therefore, reactivation of HBV can only explain the cause of liver dysfunction in a portion of the cases. Another possibility is that an improved immune system due to TB infection control may lead to an attack on the intra-hepatocyte HBV. Additionally, in HBV carriers, liver dysfunction may impair the metabolism of first-line anti-TB drugs, resulting in the accumulation of more toxic metabolites. In addition to less liver reserve in HBV carriers, these metabolites may easily further damage the liver. Furthermore, patients with chronic hepatitis B infection may have an upregulation of cytokines and a mixed inflammatory response. This pro-inflammatory condition triggered by replicating HBV may increase the susceptibility to toxic metabolites from first-line anti-TB drugs. However, the true mechanism and interaction of HBV and first-line anti-TB drugs in hepatotoxicity remains unknown, which requires further basic study to elucidate [50,54,55].
![]()
WHO definition of
hepatotoxicity
Grade 1 (mild)
<2.5 times ULN (ALT 51–125 U/L)
Grade 2 (mild)
2.5–5 times ULN (ALT 126–250 U/L)
Grade 3 (moderate)
5–10 times ULN (ALT 251–500 U/L)
Grade 4 (severe)
>10 times ULN (ALT > 500 U/L)
ALT: Alanine Aminotransferase; ULN: Upper Limit of Normal, i.e. 50 U/L
Table 1: Definition of hepatotoxicity according to the WHO Adverse Drug Reaction Terminology.
![]()
Proportion FATDH (%)
Study
Reference
8.7
Korea
17
2.55-71.59%
China
16
2.5
Indian
18
9.9
South Africa
20
8-15
Ethiopia
21,22,23
Table 2: Incidence of first line ant-TB drug induced hepatotoxicity in different countries.
![]()
First line drugs
Proportion FATDH (%)
Reference
Isoniazid
0.1-20
25
Rifampicin
0.6-2.7
26
Pyrazinamide
0.42
24
Ethambutol
0
26
Streptomycin
0
26
Table 3: Incidence of Hepatotoxicity attributable to a specific first-line anti -tuberculosis drugs.
Compared to TB patients without a viral hepatitis report, the percentage of HCV-positive TB patients who passed away before their TB therapy was finished was higher (21% vs. 9%) [56]. There has been worry that the danger of first-line anti-TB drug-induced hepatotoxicity is increased in patients with underlying chronic liver disease brought on by viral hepatitis. According to a Georgia study, during the six months of first-line anti-TB therapy, 18.8% of participants with normal baseline ALT levels experienced hepatotoxicity. This suggests a significant increase from baseline ALT levels. 43.8% of individuals with co-infection with HCV experienced hepatotoxicity [57].
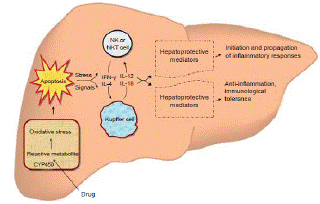
Figure 1: Pathogenesis of liver injury caused by most of the important firstline
anti-TB drugs.
Source: Kishore PV, et al. Drug-induced hepatitis with antitubercular
chemotherapy: challenges and difficulties in treatment, Kathmandu Univ
Med J. 2007;5(2):256-260.
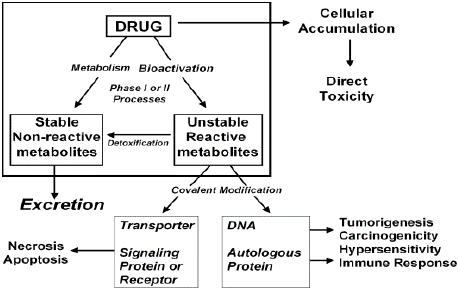
Figure 2: Proposed role of bioactivation in drug toxicity.
Source: Kalgutkar A.S., Role of Bioactivation in Idiosyncratic Drug Toxicity:
Structure–Toxicity, Relationships Advances in Bioactivation Research 2008
American Association of Pharmaceutical Scientists, DOI: 10.1007/978-0-
387-77300-1_2,
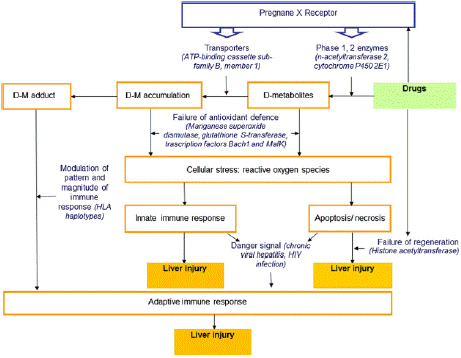
Figure 3: Hypothetical model of DILI due to first line anti-TB agents with
potential drug and host related factors (in blue) involved in the pathogenesis.
Source: Ramappa V and Aithal G, Hepatotoxicity Related to Anti-
Tuberculosis Drugs: Mechanisms and Management, Journal of Clinical and
Experimental Hepatology | March 2013 | Vol. 3 | No. 1 | 37–49
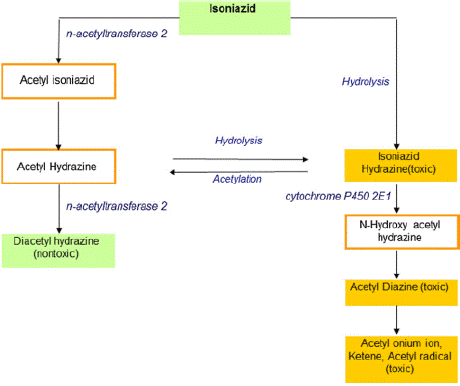
Figure 4: Pathways involved in the metabolism of isoniazid.
Source: Tostmann A .et al, Anti-tuberculosis drug-induced hepatotoxicity:
Concise up-to-date review Journal of Gastroenterology and Hepatology
(2008) 23;192–202
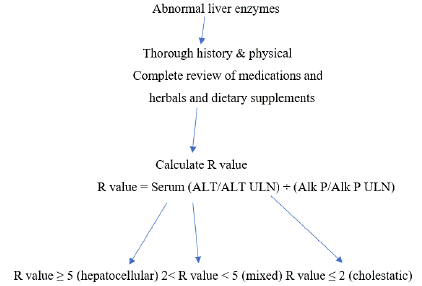
Figure 5: causality assessment and diagnosis drug induced liver injury.
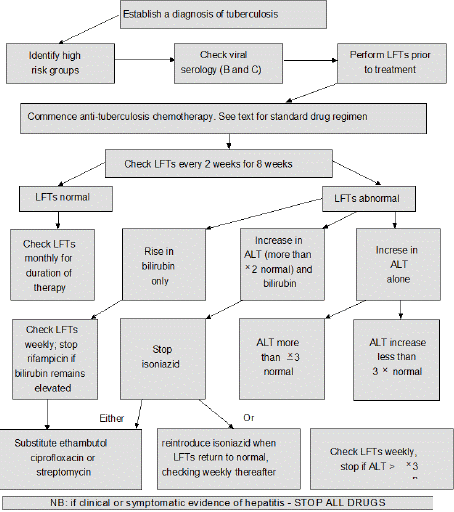
Figure 6: Protocol for anti-tuberculosis drugs and possible hepatotoxicity.
LFTs: liver function tests; ALT: alanine aminotransferase.
Source: Thompson N.et al Anti-tuberculosis medication and
the liver: dangers and recommendations in management: DOI:
10.1183/09031936.95.08081384.
One study examined the effect of HCV infection on DILI in 128 Florida inpatients receiving TB illness therapy. All had not taken alcohol or illicit drugs for at least ten days before to beginning anti-TB treatment, and they had all received isoniazid, rifampin or rifabutin, or pyrazinamide for at least five days. Hepatotoxicity affected over 30% of people with hepatitis C infection, compared to 11% of people without the virus. The development of hepatotoxicity was found to be independently correlated with hepatitis C, which increased the probability of transaminase elevation of at least 120 U/L or serum bilirubin of at least 1.5 mg/dl by five times. The chance of developing hepatotoxicity increased by more than 14 times when co-infected with HIV and hepatitis C [7].
Regarding the Egyptian study, the prevalence of HCV in TB patients was 17.02% for group I (patients with co-infection of HCV and TB) and group II (TB patients without HCV infection). In group I, six individuals (40%) had temporary transaminase increases and six cases (40%) developed DIH. There was a highly significant difference between the two groups: only 2 (3.78%) cases in group II had DIH, while 11 (20.75%) cases experienced transitory transaminase increases. In group I, there were four cases of mild DIH, one case of moderate DIH, and one case of severe DIH. Group II did not have any cases with severe DIH. ALP and total bilirubin levels, low body mass index (BMI), age ≥40, and high baseline transaminases were the risk factors for developing DIH following anti-tuberculosis medication. The first four weeks after beginning anti-tuberculosis therapy were when the majority of DIH cases (66.7% and 50% in groups I and II, respectively) occurred [58].
Alpha-interferon has recently been suggested as the first-line treatment for hepatitis C infected patients exhibiting pathologic signs of active inflammation. Alpha-interferon is thought to play a therapeutic function in individuals with co-infections of TB and HCV who experience high liver transaminase levels and pathologic indications of active inflammation, which may be a sign of HCVcaused damage. When patients with HCV infection encounter repeated increased transaminase levels when using first line anti-TB treatment, alpha-interferon may be able to facilitate the reintroduction of these drugs. Hepatitis C infection is an independent and additive risk factor for the development of DIH while receiving tuberculosis treatment [59].
It is unclear why First-line Anti-TB Drug-Induced Liver Injury (FATDILI) is more common in patients with chronic Hepatitis C Virus (CHCV) infection. Nonetheless, a number of justifications have been put up. First, as demonstrated by the aforementioned research, liver injury may be a result of HCV reactivation during hepatitis flare-ups, which can present as the presence of elevated serum HCVRNA. Second, first-line anti-TB medications may not be disposed of well in HCV carriers due to possible liver damage. This could lead to an accumulation of more hazardous metabolites. Third, the HCV core protein may alter lipid metabolism and cause steatosis. Steatosis may then compound the liver injury by interacting with DILI. When CHCV carriers receive first-line anti-TB medication, an increase in liver enzymes could indicate either DILI or CHCV activation. Generally speaking, the presence and titer of serum HCV RNA can indicate the virus's reproduction activity. In HCV carriers, HCVRNA is a more reliable marker of FATDILI than anti-HCV. But more research is needed to fully understand the actual mechanism and interactions between HCV and FATDILI [14,60].
Diagnosis and Causality Assessment in Drug Induced Liver Injury
A. History & physical examination, blood tests, hepatobiliary imaging and liver biopsy.
History and physical examination. Accurate history of medication exposure, onset, and course of liver biochemistry abnormalities is crucial. blood tests and imaging studies. The diagnostic approach to DILI can be tailored according to the pattern of liver injury at presentation. The R-value is defined as serum alanine aminotransferase / Upper Limit of Normal (ULN) divided by serum alkaline phosphatase / ULN. By common convention, R ≥ 5 is labeled as hepatocellular DILI, R < 2 is labeled as cholestatic DILI, and 2 < R < 5 is labeled as “mixed” DILI. The pattern of liver injury provides a useful framework to allow one to focus on differential diagnosis and further evaluation. However, the same medication can present with varying laboratory profiles and clinical features in individual DILI patients. Liver biopsy is not mandatory in the evaluation of DILI. Presumably, cases of less severe injury will have an even lower biopsy rate. Nevertheless, biopsy findings can be helpful and even diagnostic in some cases of suspected DILI [61].
B. Liver Fibrosis Index Score (FIB-4 score)
FBI-4 is a promising tool for liver damage evaluation. Liver fibrosis index, which is a formula that is calculated based on patients age, level of ALT and/or AST, and platelet level. Using a lower cutoff value of 1.45, a FIB-4 score <1.45 had a negative predictive value of 90% for advanced fibrosis. In contrast, FIB-4 >3.25 would have a 97% specificity and a positive predictive value of 65% for advanced fibrosis. On the definition of elevated ALT and AST levels, and this may be explained because the FIB-4 score includes age, which is considered as an important independent risk factor for DILI, and also platelet, which is also an indicator for liver function. The presence of significant fibrosis in chronic viral hepatitis indicates the need for antiviral therapies, and the outcome of therapy should be assessed by the improvement in fibrosis stage. FIB-4 index is a simple, accurate, and inexpensive method for assessing liver fibrosis [62,63].
Prevention and Management
Prevention
Liver-protecting drugs could relieve the liver dysfunction caused by first-line anti-TB medicines by getting rid of the toxin in the liver and improving the repair and regeneration of liver cell membranes. Some non-toxic herbs have activities in the form of membrane stabilizing, anti-oxidative, and CYP2E1 inhibitory effects, a reduction in lipid peroxide content in tissue and an increase in superoxide dismutase, catalase, glutathione S- transferase, and glutathione peroxidase activities should help to maintain liver cell integrity and control the increase in level of liver enzymes. The literature points to the hepato-protective effects of some synthetic compounds, such as N-acetyl cysteine, reamberine, remaxol, and ademethionine [64,65]. At the same time, a group of naturally occurring compounds has been reported as potential hepato-protective against the toxic effects of first line anti-TB drugs such as silymarin, curcumin and resveratrol [66,67].
Silymarin: A standard plant extract with strong antioxidant activity obtained from S. marianum is known to be an effective agent for liver protection and liver regeneration. Treatment of rats with INH+RIF or INH+RIF+PZA induced hepatotoxicity, as evidenced by biochemical measurements. The activities of ALT, AST, and ALP and the levels of total bilirubin were elevated, and the levels of albumin and total protein were decreased in drug-treated animals. Histopathological changes were also observed in the livers of animals that received drugs. Simultaneous administration of silymarin significantly decreased the biochemical and histological changes induced by the drugs [68].
Herbal formulation: The herbal formulation prevented hepatotoxicity significantly and improved the disease outcome as well as patient compliance without any toxicity or side effects. Curcuma Longa (CL) and Tinospora Cordifolia (TC) given as an adjuvant to standard first-line anti-TB treatment to any kind of TB patients prevented hepatotoxicity very significantly in terms of incidence, duration, and severity and also helped improve outcome in terms of quicker and more efficient achievement of sputum negativity in open, potentially infectious cases; better response in parenchymal lesion resolution; and helped improve patient compliance. Treated cases had better weight gain and significantly more reduction in Erythrocyte Sedimentation Rate (ESR) [69]. Animals treated with the mixture of triterpenic acids exhibited significantly decreased aspartate transaminase and alanine aminotransferase levels and amelioration of the histopathological alterations produced by the anti-TB drugs. The triterpene mixture was able to prevent the steatosis induced by firstline anti-TB drugs [70].
Management
Recommendations from the American Thoracic Society, the British Thoracic Society, and more recently by the National Institute for Clinical Excellence (NICE), UK, for the assessment of hepatotoxicity have been published. Risk Stratification: Pretreatment evaluation, whenever feasible, should include screening for existing chronic liver disease. Baseline evaluation should also include serology for chronic viral infections and appropriate assessment for underlying liver disease. Monitoring: Regular clinical review of patients is helpful to monitor treatment adherence, and Directly Observed Short-Course Therapy (DOTS) enhances its effectiveness. Co-administration of N-Acetyl Cysteine (NAC) was protective against DILI in animals treated with hepatotoxic doses of INH and rifampicin.
A report on the benefit of oral administration of NAC (600 mg twice daily) in the prevention of first-line anti-TB DILI comes from an open labeled trial including 60 patients of age >60 years; in the study, 37.5% developed elevation of both AST (mean 99.4) and ALT (mean 65.8) and mean bilirubin of 1.1 mg/dl within a mean duration of 5 days of starting combination anti-TB therapy when not treated with NAC [8,37].
Choice of Drugs and Regimen: Guidelines from professional bodies provide advice on the choice of drugs, combinations, and duration of therapy that include cost, affordability, access, as well as efficacy and associated adverse effects. Patient Education: Patients should be educated about the importance of adherence to medications, follow-up visits for monitoring, and symptoms of hepatotoxicity with appropriate reminders wherever possible. Interventions: Prompt withdrawal of the offending medication is the most critical intervention in the management of hepatotoxicity. If the diagnosis is drug-induced hepatitis, the anti-tuberculosis drugs should be stopped, and the drugs must be withheld until the normalization of the liver function tests. After the thorough determination of transaminases, most first-line anti-tubercular medications can be carefully restarted [71].
Conclusions and Recommendations
One typical side effect of first-line anti-TB medication is hepatotoxicity. drugs or their metabolites are extremely harmful to hepatocytes because the enzymes involved in drug metabolism in the hepatocyte microsomes may be congenital abnormalities, malformed, have low activity, or be blocked by drugs. Acute hepatic failure, drug resistance, and TB therapy cessation are frequently the outcomes of liver damage brought on by first-line anti-TB medications. The medications pyrazinamide, isoniazid, and rifampicin have the potential to be hepatotoxic.
Ethambutol and streptomycin have not been reported to cause hepatotoxicity. When compared to monotherapy used for TB prophylaxis, the use of multidrug combinations for TB treatment has been linked to an increased prevalence of DIH. Compared to patients using rapid acetylators, patients using slow acetylators are more likely to experience severe hepatotoxicity and are therefore at risk for first-line anti-TB DIH. The presence of the hepatitis B and C viruses increases the likelihood of developing DIH when receiving TB treatment.
In order to fully understand the true mechanism and interaction of HBV, HCV, and first-line anti-TB medications in hepatotoxicity, more fundamental research is necessary. It is imperative to look for hepato-protective substances that do not cause damage when used with anti-tubercular drugs. When treating patients with HBV and HCV, physicians should take into account the medication safety profiles of first-line anti-tubercular medications. When a patient's ALT elevated, liver protectors should be administered. The management and treatment of TB patients benefit from early identification and detection of hepatitis virus infection. Thus, while initiating first-line anti-TB medication for TB patients, it is imperative to test for HBV and HCV. For patients who are co-infected with HBV, HCV, and TB, routine monitoring of liver function and hepatitis viral load is advised during anti-TB treatment.
Author Statements
Competing Interest
All authors declare that they have no conflicts of interest.
References
- Pommerville J. Alcamo’s fundamentals of microbiology: Body systems: Jones & Bartlett Publishers. 2012.
- Global tuberculosis report 2022.Geneva: World Health Organization; 2022. Licence: CC BY-NC-SA 30 IGO.
- WHO, Initiative ST. Treatment of tuberculosis: guidelines: World Health Organization. 2010.
- Khalili H, Dashti-Khavidaki S, Rasoolinejad M, Rezaie L, Etminani M. Antituberculosis drugs related hepatotoxicity; incidence, risk factors, pattern of changes in liver enzymes and outcome. Daru. 2009; 17.
- Dufour DR, Lott JA, Nolte FS, Gretch DR, Koff RS, Seeff LB. Diagnosis and monitoring of hepatic injury. I. Performance characteristics of laboratory tests. Clinical chem istry. 2000; 46: 2027-2049.
- Yee D, Valiquette C, Pelletier M, Parisien I, Rocher I, Menzies D. Incidence of serious side effects from first-line antituberculosis drugs among patients treated The International Journal of Tuberculosis and Lung Disease for active tuberculosis. American journal of respiratory and critical care medicine. 2003; 167: 1472-1477.
- Saukkonen JJ, Cohn DL, Jasmer RM, Schenker S, Jereb JA, Nolan CM, et al. An official ATS statement: hepatotoxicity of antituberculosis therapy. American journal of respiratory and critical care medicine. 2006; 174: 935-952.
- Tostmann A, Boeree MJ, Aarnoutse RE, De Lange WCM, Van Der Ven AJAM, Dekhuijzen R. Antituberculosis drug induced hepatotoxicity: concise up todate review. Journal of gastroenterology and hepatology. 2008; 23: 192-202.
- Fernandez-Villar A, Sopena B, Fernandez-Villar J, Vazquez-Gallardo R, Ulloa F, Leiro V, et al. The influence of risk factors on the severity of antituberculosis drug-induced hepatotoxicity. 2004; 8: 1499-1505.
- Singla R, Sharma SK, Mohan A, Makharia G, Sreenivas V, Jha B, et al. Evaluation of risk factors for antituberculosis treatment induced hepatotoxicity. Indian Journal of Medical Research. 2010; 132.
- Lavanchy D. The global burden of hepatitis C. Liver International. 2009; 29: 74-81.
- Ghany MG, Strader DB, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009; 49: 1335-1374.
- Jawetz E, Melnick JL, Adelberg EA. Jawetz, Melnick & Adelberg’s medical microbiology: Appleton & Lange. 1995.
- Mo P, Zhu Q, Teter C, Yang R, Deng L, Yan Y, et al. Prevalence, druginduced hepatotoxicity, and mortality among patients multi-infected with HIV, tuberculosis, and hepatitis virus. International Journal of Infectious Diseases. 2014; 28: 95-100.
- Chen L, Bao D, Gu L, Gu Y, Zhou L, Gao Z, et al. Co-infection with hepatitis B virus among tuberculosis patients is associated with poor outcomes during anti-tuberculosis treatment. BMC infectious diseases. 2018; 18: 295.
- Shang P, Xia Y, Liu F, Wang X, Yuan Y, Hu D, et al. Incidence, clinical features and impact on anti-tuberculosis treatment of anti-tuberculosis drug induced liver injury (ATLI) in China. PloS one. 2011; 6: e21836.
- Jeong I, Park JS, Cho YJ, Yoon HII, Song J, Lee C-T, et al. Drug-induced hepatotoxicity of anti-tuberculosis drugs and their serum levels. Journal of Korean medical science. 2015; 30: 167-172.
- Yadav S, Pillai KK, Kapur P. Causality Assessment of Suspected Adverse Drug Reaction with Anti-Tubercular Therapy by WHO Probability Scale. Journal of Applied Pharmaceutical Science. 2011; 1: 26-29.
- Donald PR. Antituberculosis drug-induced hepatotoxicity in children. Pediatric reports. 2011; 3.
- Yimer G, Gry M, Amogne W, Makonnen E, Habtewold A, Petros Z, et al. Evaluation of patterns of liver toxicity in patients on antiretroviral and antituberculosis drugs: a prospective four arm observational study in ethiopian patients. PloS one. 2014; 9: e94271.
- Ali AH, Belachew T, Yami A, Ayen WY. Anti-tuberculosis drug induced hepatotoxicity among TB/HIV co-infected patients at Jimma University Hospital, Ethiopia: nested case-control study. PloS one. 2013; 8: e64622.
- Abera W, Cheneke W, Abebe G. Incidence of antituberculosis-drug-induced hepatotoxicity and associated risk factors among tuberculosis patients in Dawro Zone, South Ethiopia: A cohort study. International journal of mycobacteriology. 2016; 5: 14-20.
- Jeong I, Park JS, Cho YJ, II Yoon H, Song J, Lee CT, et al. Drug induced hepatotoxicity of antituberculosis drugs and their serum levels. Chest. 2011; 140: 771A.
- Cillo U, Bassanello M, Vitale A, D’Antiga L, Zanus G, Brolese A, et al. Isoniazid-related fulminant hepatic failure in a child: assessment of the native liver’s early regeneration after auxiliary partial orthotopic liver transplantation. Transplant international. 2005; 17: 713-716.
- Kishore PV, Palaian S, Paudel R, Mishra P, Prabhu M, Shankar P. Drug induced hepatitis with anti-tubercular chemotherapy: Challenges and difficulties in treatment. Kathmandu Univ Med J. 2007; 5: 256-260.
- CDC, Prevention, Society AT. Update: adverse event data and revised American Thoracic Society/CDC recommendations against the use of rifampin and pyrazinamide for treatment of latent tuberculosis infection-- United States, 2003. MMWR Morbidity and mortality weekly report. 2003; 52: 735.
- Kaplowitz N, editor Biochemical and cellular mechanisms of toxic liver injury. Seminars in liver disease; 2002: Thieme Medical Publishers, Inc., 333 Seventh Avenue, New.
- Sarich TC, Zhou T, Adams SP, Bain AI, Wall RA, Wright JM. A model of isoniazid-induced hepatotoxicity in rabbits. Journal of pharmacological and toxicological methods. 1995; 34: 109-116.
- CDC, Prevention. Fatal and severe hepatitis associated with rifampin and pyrazinamide for the treatment of latent tuberculosis infection--New York and Georgia, 2000. MMWR Morbidity and mortality weekly report. 2001; 50: 289.
- Tost JR, Vidal R, Cayla J, Diaz-Cabanela D, Jimenez A, Broquetas JM. Severe hepatotoxicity due to anti-tuberculosis drugs in Spain. The International Journal of Tuberculosis and Lung Disease. 2005; 9: 534-540.
- Sharifzadeh M, Rasoulinejad M, Valipour F, Nouraie M, Vaziri S. Evaluation of patient-related factors associated with causality, preventability, predictability and severity of hepatotoxicity during antituberclosis treatment. Pharmacological research. 2005; 51: 353-358.
- Murray KF, Hadzic N, Wirth S, Bassett M, Kelly D. Drug-related hepatotoxicity and acute liver failure. Journal of pediatric gastroenterology and nutrition. 2008; 47: 395-405.
- Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicological sciences. 2002; 65: 166-176.
- Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007; 76: 391-396.
- Park BK, Kitteringham NR, Maggs JL, Pirmohamed M, Williams DP. The role of metabolic activation in drug-induced hepatotoxicity. Annu Rev Pharmacol Toxicol. 2005; 45: 177-202.
- Lee J, BOYER J, editors. Molecular alterations in hepatocyte transport mechanisms in acquired cholestatic liver disorders. Seminars in liver disease; 2000: Thieme Medical Publishers, Inc., 333 Seventh Avenue, New.
- Ramappa V, Aithal GP. Hepatotoxicity related to anti-tuberculosis drugs: mechanisms and management. Journal of clinical and experimental hepatology. 2013; 3: 37-49.
- Huang YS, Chern HD, Su WJ, Wu JC, Chang SC, Chiang CH, et al. Cytochrome P450 2E1 genotype and the susceptibility to antituberculosis drug induced hepatitis. Hepatology. 2003; 37: 924-930.
- Metushi IG, Cai P, Zhu X, Nakagawa T, Uetrecht JP. A fresh look at the mechanism of isoniazid induced hepatotoxicity. Clinical Pharmacology & Therapeutics. 2011; 89: 911-914.
- Nakajima A, Fukami T, Kobayashi Y, Watanabe A, Nakajima M, Yokoi T. Human arylacetamide deacetylase is responsible for deacetylation of rifamycins: rifampicin, rifabutin, and rifapentine. Biochemical pharmacology. 2011; 82: 1747-1756.
- Nannelli A, Chirulli V, Longo V, Gervasi PG. Expression and induction by rifampicin of CAR-and PXR-regulated CYP2B and CYP3A in liver, kidney and airways of pig. Toxicology. 2008; 252: 105-112.
- Burk O, Koch I, Raucy J, Hustert E, Eichelbaum M, Brockmöller J, et al. The induction of cytochrome P450 3A5 (CYP3A5) in the human liver and intestine is mediated by the xenobiotic sensors pregnane X receptor (PXR) and constitutively activated receptor (CAR). Journal of Biological Chemistry. 2004; 279: 38379-38385.
- Shibata K, Fukuwatari T, Sugimoto E. Effects of dietary pyrazinamide, an antituberculosis agent, on the metabolism of tryptophan to niacin and of tryptophan to serotonin in rats. Bioscience, biotechnology, and biochemistry. 2001; 65: 1339-4136.
- Chitturi S, Farrell GC. Drug induced liver disease. Schiff’s diseases of the liver. 2011: 703-783.
- Van Hest R, Baars H, Kik S, van Gerven P, Trompenaars MC, Kalisvaart N, et al. Hepatotoxicity of rifampin-pyrazinamide and isoniazid preventive therapy and tuberculosis treatment. Clinical infectious diseases. 2004; 39: 488-496.
- Marzuki OA, Fauzi ARM, Ayoub S, Imran MK. Prevalence and risk factors of anti-tuberculosis drug-induced hepatitis in Malaysia. Singapore medical journal. 2008; 49: 688.
- Liu YM, Cheng YJ, Li YL, Liu CE, Hsu WH. Antituberculosis treatment and hepatotoxicity in patients with chronic viral hepatitis. Lung. 2014; 192: 205- 210.
- Kwon YS, Koh WJ, Suh GY, Chung MP, Kim H, Kwon OJ. Hepatitis C virus infection and hepatotoxicity during antituberculosis chemotherapy. Chest. 2007; 131: 803-808.
- Pan L, Jia ZS, Chen L, Fu EQ, Li GY. Effect of anti-tuberculosis therapy on liver function of pulmonary tuberculosis patients infected with hepatitis B virus. World Journal of Gastroenterology: WJG. 2005; 11: 2518.
- Wong WM, Wu PC, Yuen MF, Cheng CC, Yew WW, Wong PC, et al. Antituberculosis drug related liver dysfunction in chronic hepatitis B infection. Hepatology. 2000; 31: 201-206.
- Vanhoof J, Landewe S, Van Wijngaerden E, Geusens P. High incidence of hepatotoxicity of isoniazid treatment for tuberculosis chemoprophylaxis in patients with rheumatoid arthritis treated with methotrexate or sulfasalazine and anti-tumour necrosis factor inhibitors. Annals of the rheumatic diseases. 2003; 62: 1241.
- Hwang SJ, Wu JC, Lee CN, Yen FS, Lu CL, Lin TP, et al. A prospective clinical study of isoniazid rifampicin pyrazinamide induced liver injury in an area endemic for hepatitis B. Journal of gastroenterology and hepatology. 1997; 12: 87-91.
- Lee BH, Koh WJ, Choi MS, Suh GY, Chung MP, Kim H, et al. Inactive hepatitis B surface antigen carrier state and hepatotoxicity during antituberculosis chemotherapy. Chest. 2005; 127: 1304-1311.
- Wang JY, Liu CH, Hu FC, Chang HC, Liu JL, Chen JM, et al. Risk factors of hepatitis during anti-tuberculous treatment and implications of hepatitis virus load. Journal of Infection. 2011; 62: 448-455.
- Patel PA, Voigt MD. Prevalence and interaction of hepatitis B and latent tuberculosis in Vietnamese immigrants to the United States. The American journal of gastroenterology. 2002; 97: 1198-1203.
- Bushnell G, Stennis NL, Drobnik AM, Proops DC, Ahuja SD, Bornschlegel K, et al. Characteristics and TB treatment outcomes in TB patients with viral hepatitis, New York City, 2000–2010. Epidemiology & Infection. 2015; 143: 1972-1981.
- Lomtadze N, Kupreishvili L, Salakaia A, Vashakidze S, Sharvadze L, Kempker RR, et al. Hepatitis C virus co-infection increases the risk of anti-tuberculosis drug-induced hepatotoxicity among patients with pulmonary tuberculosis. PloS one. 2013; 8: e83892.
- Agha MA, El-Mahalawy II, Seleem HM, Helwa MA. Prevalence of hepatitis C virus in patients with tuberculosis and its impact in the incidence of anti-tuberculosis drugs induced hepatotoxicity. Egyptian Journal of Chest Diseases and Tuberculosis. 2015; 64: 91-96.
- Ungo JR, Jones D, Ashkin D, Hollender ES, Bernstein D, Albanese AP, et al. Antituberculosis drug–induced hepatotoxicity: the role of hepatitis C virus and the human immunodeficiency virus. American journal of respiratory and critical care medicine. 1998; 157: 1871-1876.
- Chang TE, Huang YS, Chang CH, Perng CL, Huang YH, Hou MC. The susceptibility of anti-tuberculosis drug-induced liver injury and chronic hepatitis C infection: A systematic review and meta-analysis. Journal of the Chinese Medical Association. 2018; 81: 111-118.
- Chalasani NP, Hayashi PH, Bonkovsky HL, Navarro VJ, Lee WM, Fontana RJ. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. The American journal of gastroenterology. 2014; 109: 950.
- Sterling RK, Lissen E, Clumeck N, Sola R, Correa MC, Montaner J, et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology. 2006; 43: 1317-1325.
- Vallet‐Pichard A, Mallet V, Nalpas B, Verkarre V, Nalpas A, Dhalluin‐Venier V, et al. FIB‐4: an inexpensive and accurate marker of fibrosis in HCV infection. comparison with liver biopsy and fibrotest. Hepatology. 2007; 46: 32-36.
- Baniasadi S, Eftekhari P, Tabarsi P, Fahimi F, Raoufy MR, Masjedi MR, et al. Protective effect of N-acetylcysteine on antituberculosis drug-induced hepatotoxicity. European journal of gastroenterology & hepatology. 2010; 22: 1235-1238.
- Sukhanov DS, Pavlova MV, Iablonskiĭ PK, Vinogradova TI. Comparative efficacy of clinical use of reamberin, remaxol and ademethionine in patients with tuberculosis of the respiratory organs and liver drug-injury. Antibiotics and chemoterapy 2013; 58: 13-18.
- Singh M, Sasi P, Gupta VH, Rai G, Amarapurkar DN, Wangikar PP. Protective effect of curcumin, silymarin and N-acetylcysteine on antitubercular druginduced hepatotoxicity assessed in an in vitro model. Human & experimental toxicology. 2012; 31: 788-797.
- Nicoletti NF, Rodrigues-Junior V, Santos Jr AA, Leite CE, Dias ACO, Batista Jr EL, et al. Protective effects of resveratrol on hepatotoxicity induced by isoniazid and rifampicin via SIRT1 modulation. Journal of natural products. 2014; 77: 2190-2195.
- Eminzade S, Uraz F, Izzettin FV. Silymarin protects liver against toxic effects of anti-tuberculosis drugs in experimental animals. Nutrition & Metabolism. 2008; 5: 18.
- Adhvaryu MR, Reddy NM, Vakharia BC. Prevention of hepatotoxicity due to anti tuberculosis treatment: a novel integrative approach. World Journal of Gastroenterology: WJG. 2008; 14: 4753.
- Gutiérrez-Rebolledo GA, Siordia-Reyes AG, Meckes-Fischer M, Jiménez- Arellanes A. Hepatoprotective properties of oleanolic and ursolic acids in antitubercular drug-induced liver damage. Asian Pacific journal of tropical medicine. 2016; 9: 644-651.
- Babalik A, Arda H, Bakirci N, Agca S, Oruc K, Kiziltas S, et al. Management of and risk factors related to hepatotoxicity during tuberculosis treatment. Tuberk Toraks. 2012; 60: 136-144.