
Case Report
Ann Hematol Oncol. 2017; 4(1): 1129.
Acute Promyelocytic Leukemia with Cryptic PML-RARA Translocation and Del(9q) with CD56 Positivity: A Case Report
El Jabbour TS, Dalvi SD*, Subik MK, Homan SM and Nazeer T
Department of Pathology, Albany Medical Center, USA
*Corresponding author: Dalvi SD, Department of Pathology, Albany Medical Center, 47 New Scotland Avenue, Albany, NY 12208, USA
Received: December 22, 2016; Accepted: January 24, 2017; Published: January 27, 2017
Abstract
Acute promyelocytic leukemia (APL) is classically associated with t (15;17) and has a favorable prognosis upon immediate recognition and treatment. However, a small minority of APL cases shows cryptic translocations that are detected only by polymerase chain reaction (PCR). A high index of suspicion, based on peripheral blood or bone marrow morphology, flow cytometric data, and clinical features, is essential for the diagnosis of these cases. We suggest that in such cases with a high index of suspicion, a negative karyotype or FISH should be reflexed to PCR confirmation for detection of PML-RARA transcript. We present here a rare case of APL that is positive for PML-RARA only by PCR, hasdel(9q) as the sole cytogenetic abnormality, and is positive for CD56 on flow cytometry.
Keywords: Acute myeloid leukemia; Acute promyelocytic leukemia; PMLRARA; t(15;17)
Introduction
Acute promyelocytic leukemia (APL) is a unique subtype of acute myeloid leukemia (AML), which has been designated as AML-M3 in the old French-American-British (FAB) classification, and is currently classified by the WHO as AML with recurring genetic abnormalities (APL with PML-RARA). It makes up about 5-8% of cases of de novo AML [1], with roughly 600-800 cases seen yearly in the United States [2]. The disease is rapidly fatal if untreated, with death resulting from disseminated iintravascular coagulation (DIC) and hemorrhagic or thrombotic complications.
The presence of a specific reciprocal translocation t(15;17) (q22;q12) is the defining feature of this leukemia, that results in fusion of the promyelocytic leukemia (PML) gene on chromosome 15q22 with the retinoic acid receptor-α (RARα or RARA) gene on chromosome 17q12, resulting in a PML-RARA transcript. The translated protein product functions as a novel oncoprotein and leads to maturation arrest at the promyelocyte stage of myeloid differentiation, leading to accumulation of blasts and promyelocytes in the bone marrow. This t(15;17) is seen in approximately 98% cases of APL (classical APL) [3] and is detected by either conventional karyotyping or fluorescence in situ hybridization (FISH). A rapid response to all-trans retinoic acid (ATRA) that relieves the promyelocyte maturation arrest is a feature of these classical cases.
The vast majority of the remaining cases is negative by conventional karyotyping or FISH, but is positive for the presence of the PML-RARA transcript by RT-PCR. These cases are said to have a “cryptic” or “masked” translocation, and are still responsive to ATRA. Three isoforms of PML-RARA transcript have been described [2]:
- BCR1 (L form or long form) with breakpoint in intron 6 (45-55% cases)
- BCR2 (V form or variant form) with breakpoint in various regions of exon 6 (10% of cases)
- BCR3 (S form or short form) with breakpoint in intron 3 (40-50% of cases).
However, a minority of cases are negative for PML-RARA translocation. Rather, these cases show variant translocations involving RARA with other partner genes including PLZF, NPM, NUMA, STAT5B, ZBTB16, PRKAR1A, FIP1L1, and BCOR. These cases are referred to as APL with variant RARA translocations and are often unresponsive to ATRA [1,4].
In addition to t(15;17), other chromosomal aberrations are detected in up to 50% of APL cases [2,5]. The clinical significance of these additional aberrations is controversial, with some studies demonstrating a poorer prognosis. The most frequent aberration described is trisomy 8, seen in 17% to 46% of cases [6-8]. Other anomalies described include del(9q), del(7q), abnormalities of chromosomes 1, 3, 6, trisomy 21, and isochromosome of long arm of derivative chromosome 17 originating from the t(15;17).
Case Presentation
MS is a 45-year old lady who was admitted to our hospital after falling from a chair due to pain and weakness in her lower extremities for the preceding 2 to 3 weeks. She also complained of menorrhagia and dizziness and noticed extensive bruising on the day of admission.
Complete blood count revealed hemoglobin of 8.1 g/dL; white blood cell count of 9.7x109/L; Platelet count of 9x 109/L. Her activated partial thromboplastin time was within normal limits on presentation (25 seconds; lab control 24-32 s), but gradually increased to 47 seconds during her 4-week hospital stay. Her prothrombin time was elevated on presentation (15.6 seconds; lab control: 9.8-11.8 seconds). Her fibrinogen was markedly decreased (62 mg/dL; lab normal range: 180-350 mg/dL). Examination of the peripheral smear revealed increased immature myeloid precursors with irregular cleaved nuclei, fine chromatin, inconspicuous nucleoli and moderate cytoplasmic granularity, with only occasional Auer rods detected. Bone marrow examination revealed 87% blasts and blast equivalents with similar morphology (Figures 1,2,3 and 4).
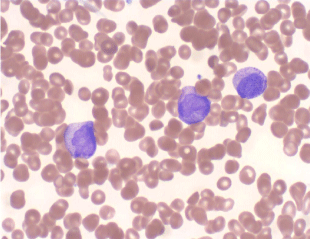
Figure 1: Peripheral blood smear shows blasts with cytoplasmic granules,
rare Auer rods and characteristic bilobed nuclei (Wright Giemsa, 50X).
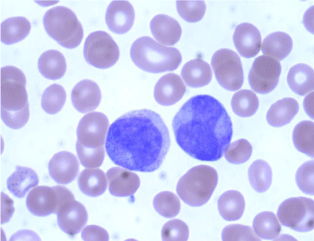
Figure 2: Peripheral blood smear showing blasts with bilobed nuclei and less
obvious cytoplasmic granules (Wright Giemsa, 100X).
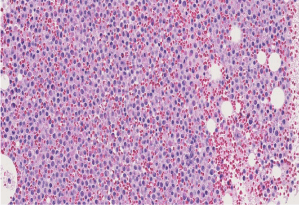
Figure 3: Bone marrow biopsy showing hypercellular marrow replaced by
myeloid blasts and equivalents (H & E, 20X).
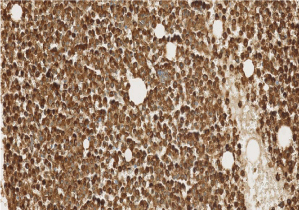
Figure 4: Bone marrow biopsy stained with myeloperoxidase immunostain
shows myeloid hyperplasia (20 X).
Flow cytometric analysis on peripheral blood revealed a 71% population of precursor myeloid cells that were positive for CD13, CD33, CD45, and myeloperoxidase, had aberrant expression of CD56, and were partially positive for CD117. They were negative for CD34, HLA-DR and lymphoid and monocytic markers (Figure 5).
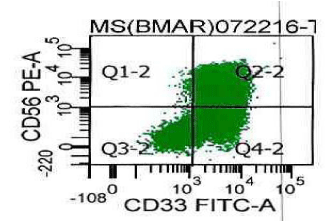
Figure 5: Flow cytometric analysis of immature myeloid cells (promyelocytes)
gated for CD45 and increased side scatter reveals the cells to be positive for
CD33 and CD56.
Notably, a head CT scan showed large areas of acute nonhemorrhagic infarction in the distribution of the right middle cerebral artery (MCA).
Thus, the morphology and immunophenotype, along with the DIC-like picture and cerebral infarction, supported a diagnosis of acute promyelocytic leukemia (APL). The peripheral blood was sent for karyotype and FISH studies to evaluate for a PML/RARA translocation.
Based on a strong suspicion for APL, the patient was started on all-trans retinoic acid (ATRA) and transfused with platelets and fresh frozen plasma.
An acute myeloid leukemia FISH panel performed on the bone marrow was negative for PML/RARA rearrangement. It was also negative for rearrangement or loss or gain of KMT2A, rearrangement involving CBFB, RUNX1T1 rearrangement, and also negative for monosomy 5, monosomy 7, and partial deletions of long arms of chromosomes 5 and 7, including CSF1R/RPS14 and MDFIC, respectively.
Due to lack of detection of PML-RARA rearrangement by FISH, the patient was started on induction chemotherapy for AML, NOS, with cytarabine and daunorubicin, however, ATRA was continued based on the strong suspicion of APL.
To further investigate for a PML-RARA transcript, the specimen was sent for quantitative RT-PCR, which was positive for a fusion of the retinoic acid receptor alpha (RARA) gene with the BCR1 (L or long form) breakpoint of the PML gene. The PCR was negative for presence of BCR2 or BCR3 transcripts (Figure 6).
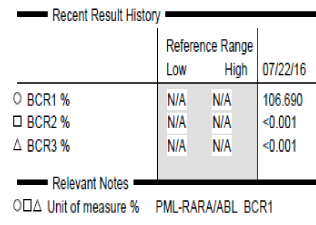
Figure 6: PCR data for PML-RARA transcript shows positive result with high
copy number for the BCR1 transcript of PML-RARA.
Interestingly, a routine karyotype performed on the bone marrow revealed a del (9)(q13q22) in 16 out of 20 metaphases analyzed. This was the sole cytogenetic abnormality identified (Figure 7).
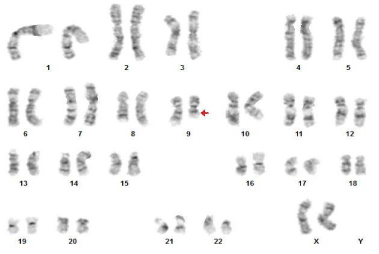
Figure 7: Karyotyping revealed del(9)(q13q22) as the sole cytogenetic
abnormality.
During admission, the patient developed fever, hypotension and pulmonary edema, attributed to APL differentiation syndrome. She also developed pancytopenia and watery diarrhea with intermittent bouts of melena, as a consequence of chemotherapy.
A repeat bone marrow performed on day 28 post therapy showed no evidence of residual acute leukemia. The patient also experienced an improvement in peripheral blood counts. Unfortunately, on day 29, the patient coded and died due to pulmonary thromboembolism.
Discussion
APL, a unique subtype of AML, has a median survival of only one month without treatment. However, current treatment protocols with ATRA alone or with the addition of arsenic trioxide are extremely effective in inducing long-lasting remissions. Hence, currently, APL has the best prognosis among the AML subtypes, with an excellent disease-free survival. Hence, prompt definitive diagnosis of this entity using a combination of conventional cytogenetics, FISH, and RT-PCR is critical. A small proportion of patients with APL may lack evidence of APL by cytogenetics and FISH; therefore, we recommend that in cases of strong clinical and morphologic suspicion, as in our present case, reflex RT-PCR testing for PML-RARA transcript be performed.
Deletion 9q has been described in 3-4% of cases of de novo AML, especially FAB M1 and M2 [9]. However, del(9q) is much rarer in APL and only few cases have been described. It is even more rare to have del(9q) as the sole karyotypic abnormality, with, to the best of our knowledge, only one case report described so far.
Our patient presented with a cerebrovascular accident and was critically ill during her hospital stay. Clinically, she responded to ATRA and her repeat bone marrow performed on day 28 showed morphologic remission. However, she died on day 29 of pulmonary thromboembolism.
Our patient was negative for PML-RARA on FISH analysis. However, her PCR was positive for the BCR1/L form of the PMLFigure RARA transcript. This suggests that she had a cryptic translocation t(15;17) that was not detectable by FISH. Also, conventional G-band karyotyping showed del(9)(q13q22) as the sole cytogenetic abnormality. This abnormality is correlated with a poor prognosis in not only APL, but also certain other subtypes of AML, notably AML with t(8;21). The presence of del(9q) as the sole cytogenetic abnormality without FISH-detectable PML-RARA fusion has been described in only one case report so far [9]. In addition, flow cytometric analysis revealed CD56 expression on the patient’s promyelocytes. CD56 positivity on promyelocytes in APL is rare and is also associated with a poor prognosis [10,11]. Hence, the concurrent presence of both del(9q) and CD56 positivity in the absence of a FISH-detectable PML-RARA translocation, in the same patient, is extremely rare, and, to the best of our knowledge, has not been previously reported. The simultaneous presence of both these poor prognostic factors was associated with an adverse clinical course for our patient, with significant morbidity and, ultimately, mortality within one month of initial diagnosis, despite appropriate therapy.
Acknowledgement
The karyotype, FISH and RT-PCR were performed at Integrated Oncology.
References
- Arber DA , Heerema-McKenney A. Acute Myeloid Leukemia, in: Hematopathology. Jaffe ES, Lee Harris N, Vardiman JW, Campo E, Arber DA. Saunders/Elsevier, Philadelphia. 2011.
- Rashidi A, Fisher SI. FISH-negative, cytogenetically cryptic acute promyelocytic leukemia. Blood Cancer J. 2015; 5: e320.
- He Y, Li X, Wang D, Hu Y, Wang W. Coexistence of cryptic insertional translocation with PML-RARa and trisomy 8 in an acute promyelocytic leukemia. J Emergency & Disaster Med. 2013; 8: 1-4.
- Wang HY, Ding J, Vasef MA, Wilson KS. A bcr3/short form PML-RARalpha transcript in an acute promyelocytic leukemia resulted from a derivative chromosome 17 due to submicroscopic insertion of the PML gene into the RARalpha locus. Am J ClinPathol. 2009; 131: 64-71.
- Wafa A, Moassass F, Liehr T, Al-Ablog A, Al-Achkar W. Acute promyelocytic leukemia with the translocation t(15;17)(q22;q21) associated with t(1;2)(q42-43; q11.2-12): a case report. J Med Case Reports. 2016; 203.
- Berger R, Le Coniat M, Derre J, Vecchione D, Jonveaux P. Cytogenetic studies in acute promyelocytic leukemia: A survey of secondary chromosomal abnormalities. Genes, Chromosomes & Cancer. 1991; 3: 332-337.
- De Botton S, Chevret S, Sanz M, Dombret H, Thomas X, Guerci A, et al. Additional chromosomal abnormalities in patients with acute promyelocytic leukemia (APL) do not confer poor prognosis: results of APL 93 trial. Br J Haematol. 2000; 111: 801-806.
- Cervera J, Montesinos P, Hernández-rivas JM, Calasanz MJ, Aventín A, Ferro MT, et al. Additional chromosome abnormalities in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and chemotherapy. Haematologica. 2010; 95: 424-431.
- Yamamoto K, Hamaguchi H, Kobayashi M, Tsurukubo Y, Nagata K. Terminal deletion of the long arm of chromosome 9 in acute promyelocytic leukemia with a cryptic PML/RARα rearrangement. Cancer Genet Cytogenet. 1999; 113: 120-125.
- Montesinos P, Rayón C, Vellenga E, Brunet S, González J, González M, et al. Clinical significance of CD56 expression in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline-based regimens. Blood. 2011; 117: 1799-1805.
- Murray CK, Estey E, Paietta E, Howard RS, Edenfield WJ, Pierce S, et al. CD56 expression in acute promyelocytic leukemia: A possible indicator of poor treatment outcome? J ClinOncol. 1999; 17: 293-297.