
Case Report
Ann Hematol Oncol. 2025; 12(3): 1482.
Acute Promyelocytic Leukemia – More than Meets the Eye
Gupta A¹, Benson R¹*, Satadeve P¹, Kachhwaha A¹, Shah B¹, Dalton P¹, Ronanki K¹, Balasubramanian P², Gupta AK², Sharma S³ and Nath UK¹
1Department of Medical Oncology Haematology, All India Institute of Medical Sciences, Rishikesh, India
2Department of Pathology and Laboratory Medicine, All India Institute of Medical Sciences, Rishikesh, India
3Consultant Cytogenetics, Core Diagnostics, Gurugram, India
*Corresponding author: Benson R, Senior Resident, Department of Medical Oncology Haematology, All India Institute of Medical Sciences, Rishikesh, India Tel: +919789097178; Email: drreshmabenson@gmail.com
Received: May 02, 2025 Accepted: June 09, 2025 Published: June 11, 2025
Keywords: Acute Promyelocytic Leukemia; ATRA; Acute Leukemia
Introduction
APL, being a unique subtype of AML in view of the morphology and molecular pathogenesis was considered a highly fatal disease with high mortality due to life threatening coagulopathy and bleeding in the early phase of the disease. Clinical presentation and morphology are the clues to diagnosis, which prompts to start therapy at the earliest suspicion, as it is considered a medical emergency. However, in rare instances, the clinician might stumble upon diagnostic cross roads, making delays and confusions in diagnosis and management. Herein, we present a series of 3 cases of AML, with similar concerns.
Case Presentations
Case 1
A 15-year-old girl presented, with history of low-grade fever for 25 days, intermittent episodes of epistaxis, bleeding per rectum and menorrhagia for 15 days, weakness of left side of the body along with aphasia for 15 days. On examination she had generalised petechial rash, pallor and left sided upper motor neuron (UMN) type of hemiparesis along with facial palsy and aphasia. She was evaluated in another center and found to have pancytopenia – hemoglobin – 8.4 gm/dL, total leucocyte count (TLC) – 540 cells/mm³ (neutrophil – 23%, lymphocytes 74 %, monocytes 4%), platelet count - 46000/mm³. Her coagulation profile was normal and peripheral smear did not reveal any atypical cells. In view of acute stroke, a Magnetic Resolution Imaging (MRI) of brain was done which showed acute infarct in right ganglio-capsulonic region, left cerebellum and left ganglio-capsulonic region along with areas of hemorrhage within. In view of pancytopenia, her bone marrow examination was done which showed 79% blasts, some of the blasts showing a polar cytoplasmic extension, giving a hand-mirror appearance (Figure 1). An immunophenotypic flow cytometric analysis revealed moderate expression of CD38, CD13, CD33, CD117, cytoplasmic MPO, CD56, while negative for CD34 AND HLA-DR - suggestive of APL. She was started on oral ATRA at 25mg/m². The Real Time Polymerase Chain Reaction (RTPCR) analysis showed PML-RARA fusion with FLT3-TKD (D8335Y/I836) positivity and Injection Arsenic Trioxide at 0.15mg/kg was added. The metaphase karyotyping showed 46 XX, t(15;17) (q24;q21). She responded clinically, her counts have improved, and is on continuous ATRA/ATO therapy at present, with antiplatelet therapy. She did not develop any differentiation syndrome during the induction therapy.
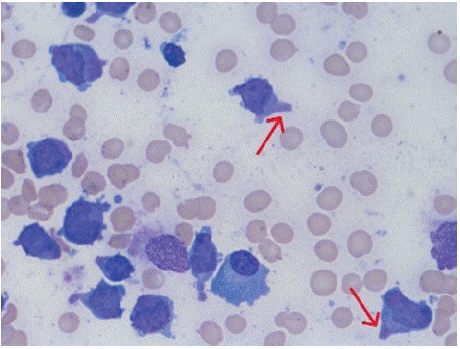
Figure 1: Bone marrow aspirate examination of case 1 under 100x showing
granular blasts with polar cytoplasmic extensions, giving a hand-mirror
appearance (red arrows).
Case 2
A 25-year-old male presented to the emergency with history of fever and myalgia for 20 days, bleeding from gums for 7 days, headache and inability to close left his left eyelid with deviation of angle of mouth to right side for 2 days. On examination, he had pallor, and gum swelling and bleeding, mild hepatosplenomegaly with left UMN 7th cranial nerve palsy and rest of the CNS examination was normal. On investigations, his hemoglobin was 4.8 g/dL, total leucocyte count was 1,43,200/mm³ (neutrophil 60%, lymphocyte 22% and monocytes 12%), and platelet count of 18000/mm³. His coagulation parameters were deranged and required blood product transfusions. His peripheral smear revealed 80% medium to large sized blasts, with high nucleocytoplasmic ratio, irregular nuclear membrane with opened up chromatin, scant to moderate amount of cytoplasm (Figure 2). Occasional blasts showed multiple auer rods (faggot cell). In view of suspected APL, ATRA at 45mg/m² was initiated immediately along with steroid prophylaxis for differentiation syndrome and hydroxyurea for cytoreduction. However, flowcytometric immunophenotypic analysis revealed blasts that showed a moderate CD38, CD64, HLADR, CD33, CD4 with negative CD13, CD14, CD34, CD117, CD11c. Based on these findings, the diagnosis was revised as acute myeloid leukemia with monocytic differentiation and ATRA and steroid was stopped. In view of poor ECOG performance status, he was initiated on low intensive chemotherapy with Injection Decitabine 20mg/m² x 5 days along with oral Venetoclax 100 mg once daily, alongwith blood product transfusions. A Computed Tomography (CT) image of brain was normal, cerebrospinal fluid (CSF) analysis revealed no atypical cells, while Contrast Enhanced (CE)MRI brain revealed subtle enhancement in mastoid part of facial nerve. In view of his CNS involvement, he also received weekly intrathecal cytarabine chemotherapy during induction. PML- RARa translocation was negative by RTPCR, while the metaphase karyotyping which followed revealed 47,XY,+X,t(11;17)(q23;q21) (Figure 3), which corresponds to the translocation resulting in RARa fusion with ZBTB16 gene. He had responded to induction therapy with day 15 induction minimal residual disease (MRD) by flow cytometry being 1%.
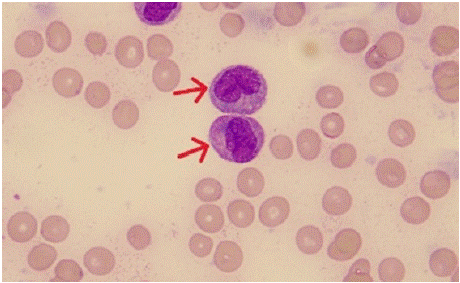
Figure 2: Peripheral smear examination of case 2 showing blasts with
high nucleocytoplasmic ratio, irregular nuclear membrane with opened up
chromatin, scant to moderate amount of cytoplasm and auer rod (red arrow).
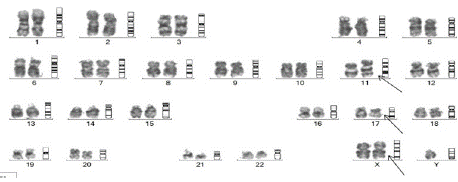
Figure 3: Metaphase karyotyping of case 2 showing 47,XY,+X,t(11;17)
(q23;q21).
Case 3
A 26-year-old male patient presented with complaints of intermittent fever and fatigue for 1months, dyspnoea for 2 days to the emergency. On examination, he had pallor, bilateral cervical level III lymphadenopathy, bilateral basal crepitations on the chest, splenomegaly of 5 cm and hepatomegaly of 1 cm below costal margin. On evaluation, his hemoglobin was 5.2 g/dl, TLC was 50700/mm³ and platelet was 7000/mm³, with derangement of coagulation parameters. His peripheral smear revealed leucocytosis with 90% blasts which were medium to large in size, have high nucleo-cytoplasmic ratio, round to slightly convoluted nuclei, prominent nucleoli and moderate amount of granular cytoplasm, with some blasts showing auer rods – suggestive of APL. He was started on ATRA at 45 mg/m², and Hydroxyurea and prednisolone to prevent differentiation syndrome. He also received blood product transfusion to correct coagulopathy. Flow cytometric analysis revealed 88.61% abnormal cells positive for CD13, CD33, CD117, cytoplasmic MPO with negative CD34 & HLADR - confirming the diagnosis of APL. Injection ATO at 0.15mg/kg was also initiated. He also received 2 doses of injection Daunorubicin on day 3 and day 5. On day 3, the patient also developed severe differentiation syndrome, for which inj dexamethasone 10 mg iv twice daily was started and required non-invasive ventilation support. However, PML – RARA fusion was negative by RTPCR, hence we performed the FISH for variant APL, which was positive. His cytogenetics report which later followed, was 46,XY, t(1;17) (q42;q21), which showed a balanced reciprocal translocation between the chromosomes 1 and 17, between the regions q42 and q21 (Figure 4), corresponding to IRF2BP2-RARA in all the metaphases analysed, which is a rare variant APL, which is sensitive to ATRA/ATO based therapy. The patient is being continued on ATRA/ATO based therapy and his post induction marrow is in morphological remission.
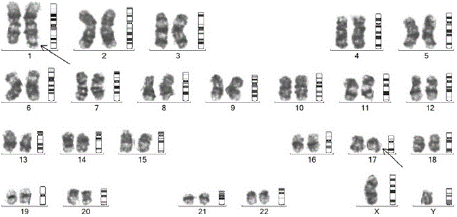
Figure 4: Metaphase karyotyping of case 3 showing 46,XY,t(1;17)(q42;q21).
Discussion
APL is a particularly aggressive subtype of AML, comprising approximately 10% of AML cases. APL has a distinct morphology and clinical presentation that may be associated with a high early death rate due to potentially fatal coagulopathy. The early death in APL has been upto 30% in some studies. Hence, once a diagnosis of APL is suspected, the disease should be managed as a medical emergency [1]. A very high index of clinical suspicion to recognise the clinical and pathological features of the disease is necessary to prevent the early mortality in APL, by initiation of ATRA and optimal management of coagulopathy [2].
Gill et al in their study has shown that initiation of ATRA after 24 hours of diagnosis was associated with a worser 30 day survival [3]. According to the National Comprehensive Cancer Network (NCCN) Guidelines, ATRA should be started before genetic confirmation in patients with clinical and pathological features of APL because early initiation of ATRA may prevent the lethal complication of bleeding resulting from disseminated intravascular coagulation [4].
The distinct changes in APL leading to coagulation abnormalities, which may manifest as hemorrhage, thrombosis or both is a very crucial clinical clue to the hematologists. The second step of suspicion is the immediate peripheral blood picture, showing abnormal promyelocytes with peculiar nuclear morphology, cytoplasmic granules and auer rods. Even in the age and advent of automated diagnostic analyzers, morphological examination that is human eye dependent remains to be the backbone in diagnosis. However, oftentimes, what meets the eye is often misleading or inadequate. In this case series, we present three different scenarios where the diagnosis and management of APL was challenging.
For instance, in microgranular variants, or in cases of pancytopenic blood picture, diagnosis from peripheral blood becomes baffling and needs bone marrow for diagnosis, which may unveil blasts with APL morphology. In our case 1, the bone marrow showed hand mirror morphology of blasts with cytoplasmic pseudopods, which is mostly described with lymphoid malignancies, though not specific. Case reports of hand mirror blast morphology in APL is quite rare. Upon literature review, we could find case reports by Bapal et al in 2021, Sim et al in 2004 and by Sun et al in 1991 [5-7]. This uncommon morphological finding had led to a delay in initiation of ATRA in this case, until the immunophenotypic findings were suggestive of APL. It is noteworthy and a takeaway from this case that abnormal promyelocytes can also present with hand mirror appearance.
Typical APL, which constitutes more than 95% cases, is cytogenetically characterized by a balanced reciprocal translocation between chromosomes 15 and 17, which results in the fusion between the promyelocytic leukemia (PML) gene and retinoic acid receptor-a (RARa) and is sensitive to differentiation therapy containing alltrans retinoic acid (ATRA) and arsenic trioxide (ATO). They exhibit an excellent clinical outcome even with chemotherapy free regimens. However, a rare subset of around 5% cases with morphologic, cytochemical and immunophenotypic features of APL show variant translocations involving the RARA gene at 17q21, named as variant APL [8]. Around 20 different fusion partners have been identified other than PML, in an excellent review on variant APL by Zhang et al [9]. Some of these are resistant to ATRA/ATO based therapy. The identification and understanding of these variant APLs are important in deciding the treatment regimen for optimal response.
in the second case, the clinical presentation with overt DIC and CNS involvement, was highly suggestive of APL, with the peripheral blood smear showing blasts with promyelocyte like morphology, auer rods and faggot cells. ATRA was prudently started in this case, however, the flowcytometric picture was suggestive of blasts belonging to monocytic lineage than promyelocytes. The cytogenetic analysis however revealed the presence of PLZF or ZBTB16- RARA fusion, which is the best described variant translocation of APL, which is intrinsically resistant to ATRA/ATO based therapy. Though the morphological picture in peripheral smear was suggestive of APL, it is also often confused with AML with monocytic differentiation, as in this case. As the blasts are not immunophenotypically promyelocytic, this will be a rare case of AML with t(11;17), and not APL. Upon our literature review, previously, only 4 cases have been reported with this translocation in AML – M5 [10-13].
In the third case, which was clinically and morphologically APL the negativity of PML-RARA fusion transcript contemplated a doubt of variant APL. This prompted further evaluation, to the detection of IRF2BP2-RARA fusion, which is also a rare entity. Only seven cases of IR2BP2 translocation has been reported so far in literature and this will be the eighth case reported [14-20].
Conclusion
The prompt diagnosis and opportune treatment initiation is lifesaving in APL, which converts this highly fatal disease to a highly curable one. The diagnostic dilemmas in typical and variant APLs pose a challenge to the clinicians and pathologists. Quick identification of variant APL is also required as change in treatment might be required in some variants. Through this case series, we have described three cases which posed a difficulty in diagnosis. Further studies and reports are required in the future describe to study the landscape of morphology and genetics of APL. This might also pave way for further targeted therapies in variant APLs.
Authors’ Contribution
Conceptualization: BR, GA, SB, NN;
Writing - original draft preparation: BR, GA, KA, RK;
Writing - review and editing: BR, GA, SP, DP;
Resources: BR, NN, RK, SS, BP, GAK;
Supervision: BP, GAK, NUK.
Ethics Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/ or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent has been obtained from the patients included in this case report to participate.
References
- Sanz MA, Fenaux P, Tallman MS, Estey EH, Löwenberg B, Naoe T, et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood. 2019; 133: 1630– 1643.
- Odetola O, Tallman MS. How to avoid early mortality in acute promyelocytic leukemia. Hematology. 2023; 2023: 248–253.
- Gill H, Yung Y, Chu HT, Au WY, Yip PK, Lee E, et al. Characteristics and predictors of early hospital deaths in newly diagnosed APL: a 13-year population-wide study. Blood Advances. 2021; 5: 2829–2838.
- National Comprehensive Cancer Network: Clinical Practice Guidelines in Oncology: Acute myeloid leukemia, version 2. 2025.
- Sun T, Weiss R. Hand-mirror variant of microgranular acute promyelocytic leukemia. Leukemia. 1991; 5: 266–269.
- Bapat KN, Patil RB. The Hand Mirror Image: Which is the Lineage in the End? Annals of Pathology and Laboratory Medicine. 2021; 8: C10-12.
- Sim J, E Ma. Acute Promyelocytic leukemia or not? Hong Kong Association of Blood transfusion and Hematology. 2004.
- The 5th Edition of the World Health Organization Classification of Hematolymphoid Tumors. In: Li W, editor. Leukemia. Brisbane (AU): Exon Publications, 2022.
- Zhang X, Sun J, Yu W, Jin J. Current views on the genetic landscape and management of variant acute promyelocytic leukemia. Biomarker Research. 2021; 9: 33.
- Classen CF, Teigler-Schlegel A, Röttgers S, Reinhardt D, Döhner K, Debatin KM. AML bearing the translocation t(11;17)(q23;q21): involvement of MLL and a region close to RARA, with no differentiation response to retinoic acid. Ann Hematol. 2005; 84: 774–780.
- Shekhter-Levin S, Gollin SM, Kaplan SS, Redner RL. Involvement of the MLL and RARa genes in a patient with acute monocytic leukemia with t(11;17) (q23;q12). Leukemia. 2000; 14: 520–522.
- Dubé S, Fetni R, Hazourli S, Champagne M, Lemieux N. Rearrangement of the MLL gene and a region proximal to the RARa gene in a case of acute myelocytic leukemia M5 with a t(11;17)(q23;q21). Cancer Genetics and Cytogenetics. 2003; 145: 54–59.
- Reeves BR, Kempski H, Jani K, Borrow J, Howe K, Solomon E, et al. A case of acute monocytic leukemia with t(11;17) involving a rearrangement of MLL-1 and a region proximal to the RARA gene. Cancer Genetics and Cytogenetics. 1994; 74: 50–53.
- Yin CC, Jain N, Mehrotra M, Zhagn J, Protopopov A, Zuo Z, et al. Identification of a Novel Fusion Gene, IRF2BP2-RARA, in Acute Promyelocytic Leukemia. Journal of the National Comprehensive Cancer Network. 2015; 13: 19–22.
- Mazharuddin S, Chattopadhyay A, Levy MY, Redner RL. IRF2BP2-RARA t(1;17)(q42.3;q21.2) APL blasts differentiate in response to all-trans retinoic acid. Leukemia & Lymphoma. 2018; 59: 2246–2249.
- Shimomura Y, Mitsui H, Yamashita Y, Kamae T, Kanai A, Matsui H, et al. New variant of acute promyelocytic leukemia with IRF2BP2–RARA fusion. Cancer Sci. 2016; 107: 1165–1168.
- Liu Y, Xu F, Hu H, Wen J, Su J, Zhou Q, et al. A rare case of acute promyelocytic leukemia with IRF2BP2-RARA fusion; and literature review. OncoTargets and Therapy. 2019; 12: 6157–6163.
- Schwartz JG, Clare N, Hansen K, Britton H, Manhoff L. Acute promyelocytic leukemia: report of a variant translocation, t(1;17). Cancer Genet Cytogenet. 1986; 20: 89–93.
- Alotaibi AS, Abdulrazzaq M, Patel KP, Ravandi F, Konoplev S, Bueso-Ramos C, et al. Acute promyelocytic leukemia (APL) with an IRF2BP2-RARA fusion transcript: an aggressive APL variant. Leukemia & Lymphoma. 2020; 61: 3018–3020.
- Jovanovic JV, Chillón MC, Vincent-Fabert C, Dillon R, Voisset E, Gutiérrez NC, et al. The cryptic IRF2BP2-RARA fusion transforms hematopoietic stem/ progenitor cells and induces retinoid-sensitive acute promyelocytic leukemia. Leukemia. 2017; 31: 747–751.