
Research Article
Ann Hematol Oncol. 2025; 12(1): 1471.
Severe Hemolytic Crisis in Pediatric Patients with Sickle Cell Disease without History of Recent Transfusion: A Single Center, Multi-site Case Series
Garrett ER1,2* and Rollins MR1,2
1Department of Pathology and Laboratory Medicine, Children’s Healthcare of Atlanta, Atlanta, GA, USA
2Department of Pediatrics, Division of Hematology-Oncology, Emory University SOM, Atlanta, GA, USA
*Corresponding author: Erin R. Garrett, MS, MD, 2220 N. Druid Hills Rd, 3rd Fl Blood Bank, Atlanta, GA, USA Tel: 951)642-0603; Email: Erin.Garrett@choa.org
Received: January 23, 2025; Accepted: February 11, 2025; Published: February 13, 2025
Abstract
Antibody negative Delayed Hemolytic Transfusion Reaction [Ab(-) DHTR] is defined in patients with Sickle Cell Disease (SCD) as hemolysis within 21 days following red blood cell (RBC) transfusion in the absence of positive serologic testing (direct antiglobulin test (DAT) and/or new alloantibody). Hyperhemolysis (HH), or indiscriminate hemolysis of autologous and allogeneic RBCs, is a unique complication of Ab- DHTR in patients with SCD. With a constellation of lab findings including elevated LDH, reticulocytopenia and a rapid drop in hemoglobin below pre-transfusion and baseline level, Hyper-Hemolysis Syndrome (HHS) is an important complication of Ab(-) DHTR that can result in morbidity and mortality among patients with SCD. Despite associated lab and clinical findings, diagnosis and prompt intervention can be difficult due to the underlying pathophysiology of SCD with the added context of recent transfusion.
In this retrospective case review of five pediatric patients with SCD, we describe their courses during an inpatient hospital admission for management of suspected DHTR by the clinical team. Laboratory findings were supportive of Ab (-) DHTR complicated by HH, however, the patients were subsequently found not to have transfusion exposure within the 21 days prior to presentation. Considering HH in a patient with SCD a distinct clinical presentation without temporal relation to recent transfusion should be considered to prevent delay in appropriate management. While the mechanism of action of HH/HHS remains elusive, alternative inciting events and their mechanism of actions should continue to be explored to direct therapy and improve patient outcomes for patients with SCD.
Keywords: Hyper-hemolysis; Sickle Cell Disease
Abbreviations
Ab(-)DHTR: Antibody Negative DHTR; ACS: Acute Chest Syndrome; ASH: American Society of Hematology; AUS: Activity of Undetermined clinical Significance; CDC/NHSN: Center for Disease Control and Prevention’s National Healthcare Safety Network; DAT: Direct Antiglobulin Test; DHTR: Delayed Hemolytic Transfusion Reaction; DOS: Day(s) of Stay; FDA: Food and Drug Administration; Hct: Hematocrit; Hgb: Hemoglobin; HH: Hyper-hemolysis; HHS: Hyper-hemolysis Syndrome; HU: Hydroxyurea; LDH: Lactate Dehydrogenase; LOS: Length of Stay; MOD: Multi-organ Dysfunction; MTP: Massive Transfusion Protocol; PNH: Paroxysmal Nocturnal Hemoglobinuria; SCD: Sickle Cell Disease; RBCs: Red Blood Cells; TMA: Thrombotic Microangiopathy; VOC: Vaso-Occlusive Crisis
Introduction
Sickle cell disease (SCD) is a multisystem disease associated with episodes of acute illness and progressive organ damage; it is one of the most common severe monogenic disorders worldwide [1]. Since being first described in 1910, patient life span and quality have vastly improved due to increased knowledge of disease pathophysiology and technological advancements in pharmacology and gene therapy [2]. Despite these advancements, transfusion of allogeneic RBCs remains a pillar of therapy for a specific subset of patients to treat current or mitigate future disease related complications [3]. Allogeneic RBCs do, however, have associated risks, including alloimmunization, transfusion associated hemosiderosis, transfusion transmitted infection and other transfusion related adverse events as defined by the CDC/NHSN Hemovigilance Module [4].
Previous studies have shown that patients with SCD are at increased risk of developing alloimmunization as well as experiencing other transfusion related adverse events compared to the general population; specifically, DHTR [5]. DHTR is reported more frequently in patients with SCD but is believed to be underreported due to symptomology resembling VOC [6,7].
DHTR is defined by CDC/NHSN as a rapid fall in hemoglobin (Hgb) to pre-transfusion levels with positive direct antiglobulin test (DAT), and identification of new alloantibody occurring 24 hours to 28 days following transfusion. There is near total, or complete, destruction of allogeneic RBCs sparing autologous RBCs. Patients may present with symptoms of fever, chills, pain, hemoglobinuria, and jaundice from intravascular, antibody mediated hemolysis. They can also be asymptomatic, with destruction of allogeneic RBCs and unintentional detection of new alloantibody identified by screening labs performed for an unrelated reason. Neither presentation is unique for, nor specific to, patients with SCD. Treatment is typically supportive and additional transfusion of RBCs with avoidance of antigens to newly identified or historic alloantibody (-ies) is considered safe and appropriate management for future transfusion [5].
Ab(-)DHTR has been described in the literature with clinical symptoms identical to the CDC/NHSN defined DHTR absent the newly identified alloantibody and/or positive DAT [8]. The etiology and pathophysiology of this presentation remain obscure, making its prevalence difficult to quantify. ASH has implemented management guidelines for patients with SCD presumed to have Ab(-) DHTR presenting within 21 days of transfusion with signs of hemolysis and no serological evidence of DHTR [9]. In these cases, additional transfusion is not advisable due to the lack of alloantibody identification for antigen avoidance.
A reported consequence of severe Ab(-) DHTR is the development of potentially life-threatening HH. HH is defined as rapid decline of post-transfusion HgbA level to below pre transfusion levels. The mechanism of this “bystander” hemolysis, destruction of both allogeneic and autologous RBCs, has yet to be identified. In patients with SCD, HH has been characterized as an extreme complication of Ab(-) DHTR [8]. HHS is a constellation of laboratory findings including elevated LDH and reticulocytopenia in addition to the rapid drop in Hgb below pre-transfusion and baseline associated with HH, and severe pain out of proportion to previously reported VOC. Given the underlying pathophysiology of SCD, making a definitive diagnosis of Ab- DHTR or HH/HHS can be difficult in proximity to transfusion, and attempts to develop a diagnostic algorithm have had limited utility [7,10,11]. However, a unifying theme of prior history of transfusion within 21-28 days of presentation and/or symptom onset is consistent for Ab-DHTR and HH/HHS.
In 2006, Ballas and Marcolina described a subset of adult patients with SCD presenting with clinical and laboratory features of HH/HHS manifesting as “acute painful episodes [12].” Pain out of proportion to historical VOC has been reported as an associated feature of HH/HHS clinical presentation. Since patients in the study with history of transfusion within 44 months of presentation were excluded, the authors concluded that HH/HHS in these patients was a complication of underlying disease, not proximity to transfusion. A case reported by Jones et al in 2015, describes a single pediatric patient presenting with HH following a VOC and no lifetime history of transfusion exposure [13]. These cases support an alternative to transfusion as a trigger for or association with HH/HHS, suggesting it is acute on chronic disease related hemolysis not directly related to transfusion. It is also important to consider the clinical implications of correlating this presentation specifically with recent transfusion and the impact on time to diagnosis and management of those affected patients. Herein, we describe a cohort of pediatric patients with SCD presenting with severe hemolysis and no transfusion exposure within at least 28 days of presentation.
Methods
The Institutional Review Board of a single center, multi-site tertiary care pediatric hospital in the Southeastern US, waived a retrospective chart review conducted between November 2021 and January 2023. Patient inclusion criteria were: diagnosis of SCD (any genotype), admitted for documented concern for DHTR with date of last transfusion unknown at the time of admission, and a Transfusion Medicine consult for evaluation for DHTR. Patients with a confirmed history of no transfusion documented at our institution or outside institution >28 days prior to presentation with a negative antibody screen and DAT from admission to discharge were identified.
Patient demographics including age, admission diagnosis, SCD genotype, current disease modifying agents such as HU, history of allo-/autoimmunization, and date of last transfusion. The following immunohematology, hematologic and metabolic labs results were recorded during admission: blood type, antibody screen, antibody identification, DAT, Hgb electrophoresis, Hgb, Hct, platelet count, reticulocyte count (percentage and absolute), LDH, bilirubin, haptoglobin and plasma free Hgb. Medical interventions received during admission including transfusion, medications used for management of suspected hemolysis, and apheresis were also documented.
Results
Demographics
Five patients with a median age of 17y (15-19y) and 6 distinct encounters were identified. Two patients were siblings. Patient #5 had 2 encounters with <24hrs between discharge and re-admission. Three of 5 patients were HgbSS genotype. The majority of the patients were Black females (80%). All patients had a history of prior transfusion. Two of 5 patients (40%) had a history of alloimmunization and 1/5 (20%) had a history of AUS with all clinically significant alloantibodies ruled out. No patients had a history of autoantibodies. Three of 5 patients (60%) were on HU only; the other 2 patients were treated with Voxeletor in combination with HU or Crizanlizumab. Outpatient medication compliance was not evaluated. See Table 1 for additional demographic information.
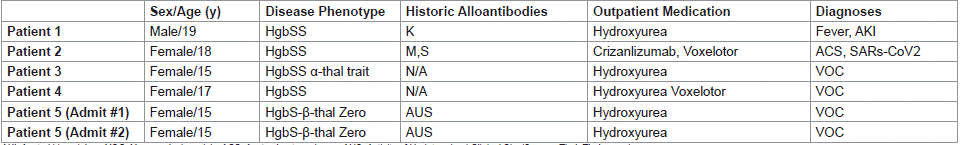
Table 1: Patient Demographics.
All patients in our cohort presented >30 days from their last transfusion (36-1311 days), with 80% presenting >1 year. One exception is encounter #2 for Patient #5. This patient was readmitted +6 days after their last transfusion which occurred during encounter #1. The documented indication for transfusion during encounter #1 was symptomatic anemia. See table #2 for additional details.
Laboratory Studies
All patients had a negative antibody screen and negative DAT test with polyspecific reagent (IgG/C3) on admission. Average Hgb at presentation was 9.7 g/dL (5.3 g/dL-10.8 g/dL), with a nadir achieved by DOS 5 for most patients. The average absolute reticulocyte count on admission was 255.3 (45.6-481.8) with nadir achieved DOS 5 (average 100.8; range 20.4-218.7). Four of 5 patients demonstrated mild (<150k) to moderate (<100k) thrombocytopenia with patient #5 exhibiting this only during encounter #2.
There was variable institutional practices in obtaining markers of hemolysis, including HgbA, and other metabolites when managing patients with suspected DHTR or HH/HHS. The results of labs including LDH, plasma free Hgb, haptogobin and total/fractionated bilirubin in addition to select complement studied can be found on Table 2. Abnormal values are bolded and italicized. Complete blood count and reticulocyte values (%, and absolute) were the only values obtain for all patients at some time point during their encounter.
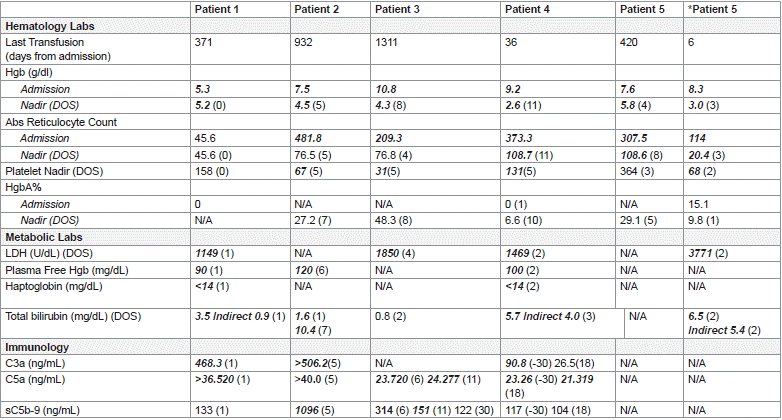
Table 2: Lab Values.
Clinical Features and Course
All patients reported symptoms of VOC including head, back, and bilateral upper/lower extremity pain. One patient demonstrated symptoms with radiographic confirmation of ACS as well as neurologic and renal impairment. Two patients were SARs- COV2 positive, with viral pneumonitis and reactive airway disease respectively on radiologic imaging. Four patients presented with leukocytosis (>11,000/uL), only one of whom was febrile.
The average LOS) for this cohort was 16 days (8-26 days). No patients received IVIg. Four of 5 patients received Erythropoietin with Patient #5 receiving it only during encounter #2. All patients in our cohort received RBC transfusion during all encounters with Patient #5 receiving it only during admission #1. Patient #5 received plasma exchange for 5 episodes during encounter #2 for suspected MOD. Patient #2 required MTP activation for management for acute cardiac arrest and was the only patient in our cohort documented with SARs-COV2. This was also the only patient in our cohort who expired prior to discharge.
Discussion
To our knowledge, this is the first pediatric case series demonstrating symptoms and laboratory evidence of HH/HHS without a temporal association to transfusion in patients with SCD. These patients and their clinical course correlate with the patient initially presented by Jones et al [13]. The current range of literature focuses on the diagnosis and management of HH/HHS in the context of recent transfusion as a severe complication of Ab-DHTR. To this extent, our case series is novel.
Patient #5 is the only patient in our cohort who presented with a history of transfusion >1 year who was transfused for severe anemia attributable to a complication of underlying SCD during encounter #1 and subsequently presented within 1 week of transfusion with worsening symptoms. It is unclear if encounter #2 complicated by MOD and HH were de novo versus a progression of encounter #1; however, VOC and anemia with reticulocytopenia were documented in encounter #1 prior to transfusion. In this instance, HH/HHS likely presented during encounter #1 prompting the transfusion, and not because of transfusion resulting in encounter #2. The subsequent severity of her clinical course was in all probability multifactorial with significant bystander contributing to an inflammatory milieu.
The presenting symptom of severe VOC out of proportion to previous VOC is reported in patients with SCD in cases of Ab- DHTR related HH/HHS [14-16]. The most common laboratory finding reported in the literature is a drop in Hgb below baseline/ pre-transfusion levels with reticulocytopenia. Elevated LDH and elevated total and/or indirect bilirubin were also reported as evidence of indiscriminant RBC hemolysis [6,8,11,12]. The degree of LDH elevation seen in our patient cohort is not comparable to that described in patients with associated DHTR, however, they are comparable to those described by Ballas and Marcolina [12]. Figure 1 compares the LDH of 4 patients in our cohort during their admission. The values for Patient #5 represent encounter #2. The peak bilirubin of Patient #5 being 4 times greater than the peak of the other recently nontransfused patients in the cohort is likely a reflection representation of the severity of bystander hemolysis from the previous transfusion and likely correlate with exposure to recent previous transfusion.
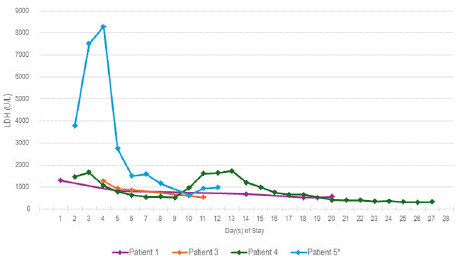
Figure 1: LDH Trends.
The complement pathway has been implicated in the development of HH/HHS. Prior studies have demonstrated hypercomplementemia at baseline in patients with SCD with the suggestion that some disease related complications such as VOC may be a result of activation of the complement pathway [17,18]. Eculizumab, a C5bC5b complement inhibitor, was approved by the FDA in 2007 for the treatment of complement mediated hemolysis including PNH and has been shown to be effective in cases of complement mediated TMA [19]. All patients in our cohort received at least one dose of Eculizumab, with Patient #5 receiving the drug only during encounter #2. Timing of dosing associated with DOS can be found in Table 3. Select complement study results can be found in Table 2.

Table 4: Medical Interventions and Disposition.
Conclusion
Here, we present a retrospective case series report of pediatric patients with SCD and severe hemolysis that meet the clinical criteria for HH/HHS but without recent transfusion as defined by ASH (<21 days prior to presentation). This suggesting there may not be a direct correlation between recent transfusion exposure and HH/HHS as a complication of Ab- DHTR. Patients with SCD have a complex pathophysiology that results in acute on chronic anemia from ongoing hemolysis resulting in inflammation with a variety of clinical and laboratory manifestations including but not limited to VOC. Studies have demonstrated that patients with SCD are in a “pro-inflammatory state” which is further exacerbated in patients presenting in an acute setting [20].
Transfusion in patients with SCD can be a lifesaving intervention when they present for management of severe complications. Unfortunately, transfusion is not risk free. In the case of Ab-DHTR or HH/HHS, as opposed to DHTR or other SCD related complications, is not recommended as an intervention for management. While Ab- DHTR is a clinical entity used to describe patients who have received transfusion and have hemolysis with no immunological findings, it may be important to reassess the temporal relationship of transfusion to a patient presenting with clinical features of HH/HHS and no recent transfusion. Considering HH/HHS as a pathophysiologic entity independent of transfusion status may result broadening the scope of potential high risk patients that may require a more acute intervention. It may be prudent to identify a constellation of labs (reticulocytopenia, relative thrombocytopenia, and severe hemolysis) and physical findings (VOC out of proportion to normal episodic pain) as part of screening for impending hyper-hemolysis, independent of transfusion exposure.
There are many limitations to this series. As a retrospective chart review, there are gaps in data points secondary to variations in clinical practice. Post hospitalization follow up was not evaluated, therefore, a full timeline to resolution of HH/HHS could not be ascertained. Parvovirus, among other viral etiologies, can result in marrow suppression and SCD symptom exacerbation. With the exception of SARs-CoV2, viral studies were not consistently obtained on any of the patients in this cohort. Therefore, its confounding effect on the patients’ presentations and ultimate outcomes could not be assessed. Lastly, there is likely selection bias unaccounted for due to the fact that Transfusion Services had to be consulted in order for the patient to be considered for this cohort. This may have inadvertently excluded patients identified as HH/HHS without recent transfusion.
We hope this case series will spark continued investigation of the pathophysiology of HH/HHS in patients with SCD to confirm. Additionally, we hope to promote further discussion on screening and managing patients with SCD that present with signs and symptoms of HH/HHS independent of the temporal relationship to transfusion.
References
- Weatherall D, Hofman K, Rodgers G, Ruffin J, Hrynkow S. A case for developing North-South partnerships for research in sickle cell disease. Blood. 2005; 105: 921-923.
- Salinas Cisneros G, Thein SL. Recent Advances in the Treatment of Sickle Cell Disease. Front Physiol, 2020; 11: 435.
- Chou ST. Transfusion Therapy for sickle cell disease: a balancing act. Hematology Am Soc Hematol Educ Program. 2013; 1: 439-446.
- Harvey AR, Basavaraju SV, Chung KW, Kuehnert MJ. Transfusion-related adverse reactions reported to the National Healthcare Safety Network Hemovigilance Module, United States, 2010 to 2012. Transfusion. 2015; 55: 709-718.
- Rollins MR, Chou ST. Adverse events of red blood cell transfusions in patients with sickle cell disease. Transfus Apher Sci. 2022; 61: 103557.
- Habibi A, Mekontso-Dessap A, Guillaud C, Michel M, Razazi K, Khellaf M, et al. Delayed hemolytic transfusion reaction in adult sickle-cell disease: presentations, outcomes, and treatments of 99 referral center episodes. Am. J. Hematol. 2016; 91: 989-994.
- Narbey D, Habibi A, Chadebech P, Mekontso-Dessap A, Khellaf M, Lelièvre JD, et al. Incidence and predictive score for delayed hemolytic transfusion reaction in adult patients with sickle cell disease. Am J Hematol. 2017; 92: 1340-1348
- Talano JA, Hillery CA, Gottschall JL, Baylerian DM, Scott JP. Delayed hemolytic transfusion reaction/hyperhemolysis syndrome in children with sickle cell disease. Pediatrics. 2003; 111: e661-665.
- Chou ST, Alsawas M, Fasano RM, Field JJ, Hendrickson JE, Howard J, et al. American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv. 2020; 4: 327-355.
- Adkins BD, Sharma D, Eichbaum Q. Can we better predict delayed hemolytic transfusion reactions and hyperhemolysis in sickle cell disease? Transfus Apher Sc. 2020; 59: 102681.
- Fasano RM, Miller MJ, Chonat S, Stowell SR. Clinical presentation of delayed hemolytic transfusion reactions and hyperhemolysis in sickle cell disease. Transfus Clin Biol. 2019; 26: 94-98.
- Ballas SK, Marcolina MJ. Hyperhemolysis during the evolution of uncomplicated acute painful episodes in patients with sickle cell anemia. Transfusion. 2006; 46: 105-110.
- Jones EA, Smith L, Keenan RD. Hyperhaemolysis in Sickle Cell Disease Is Not Necessarily a Transfusion Reaction. Blood. 2015; 126: 4595.
- Dean CL, Maier CL, Chonat S, Chang A, Carden MA, El Rassi F, et al. Challenges in the treatment and prevention of delayed hemolytic transfusion reactions with hyperhemolysis in sickle cell disease patients. Transfusion. 2019; 59: 1698-1705.
- Dumas G, Habibi A, Onimus T, Merle JC, Razazi K, Mekontso Dessap A, et al. Eculizumab salvage therapy for delayed hemolysis transfusion reaction in sickle cell disease patients. Blood. 2016; 127: 1062–1064.
- Deepika S Darbari, Onyinye Onyekwere, Mehdi Nouraie, Caterina P Minniti, Lori Luchtman-Jones, Sohail Rana, et al. Markers of Severe Vaso-Occlusive Painful Episode Frequency in Children and Adolescents with Sickle Cell Anemia. The Journal of Pediatrics. 2012; 160: 286-290.
- Roumenina LT, Chadebech P, Bodivit G, Vieira-Martins P, Grunenwald A, Boudhabhay I, et al. Complement activation in sickle cell disease: Dependence on cell density, hemolysis and modulation by hydroxyurea therapy. Am J Hematol. 2020; 95: 456– 464.
- Yoo JJ, Graciaa SH, Jones JA, Zuo Z, Arthur CM, Leong T, et al. Complement Activation during Vaso-Occlusive Pain Crisis in Pediatric Sickle Cell Disease. Blood. 2021; 138: 858.
- Chonat S, Graciaa S, Shin HS, Newton JG, Quarmyne MO, Boudreaux J, et al. Eculizumab for complement mediated thrombotic microangiopathy in sickle cell disease. Haematologica. 2020; 105: 2887-2891.
- Conran N, De Paula EV. Thromboinflammatory mechanisms in sickle cell disease - challenging the hemostatic balance. Haematologica. 2020; 105: 2380-2390.