
Research Article
Austin J Comput Biol Bioinform. 2023; 4(1): 1017.
In-Silico Characterization of Pathogenic Missense nsSNPs in AAMDC Human Gene Involved in Onset of Breast Cancer
Muhammad Ali Raza*
Department of Biological Sciences, University of Sialkot, Pakistan
*Corresponding author: Muhammad Ali Raza Department of Biological Sciences, University of Sialkot, Pakistan. Email: aliraza03440044484@gmail.com
Received: October 27, 2023 Accepted: November 28, 2023 Published: December 05, 2023
Abstract
Breast cancer is a type of cancer that occurs when cells in the breast tissue grow uncontrollably. It may be caused by activation of growth receptors and/or mutations in oncogenes. Single Nucleotide Polymorphisms (SNPs) are variations in the DNA sequence that occur when a single nucleotide in the genome is altered. Non-synonymous SNPs (nsSNPs) can alter the amino acid sequence of a protein, potentially affecting its structure, function, and interaction with other proteins. In the context of breast cancer research, in-silico nsSNPs analysis can help identify specific genetic mutations that may be associated with increased breast cancer risk or treatment response. A newly discovered gene, AAMDC, is an oncogene, mutation in which can lead to breast cancer. The purpose of this study is to find out the possible and vulnerable mutational sites in its sequence. Designed study primarily focused in-silico structural analysis and functional analysis of nsSNPs associated with AAMDC gene. 175 most damaging nsSNPs of AAMDC gene were analyzed using different bioinformatics tools. After sequence analysis and protein stability prediction using SIFT, Polyphen-2, CADD, and I-Mutant2.0, 10 nsSNPs were shortlisted using consensus based approach. All nsSNPs were analyzed for disease association prediction using IMutant2.0, PhD-SNP and PANTHER; 3 nsSNPs were shortlisted based on consensus approach. Then structural and functional variation of these damaging SNPs was analyzed using MUTPRED2 and SNAP2. The goal of current study is to check out the damaging effect of nsSNPs associated with structure and function of AAMDC gene. This study can help us in understanding breast cancer genetics and its prevention and treatment.
Keywords: Breast Cancer; nsSNPs; AAMDC; in-silico
Introduction
Cancer is characterized by uncontrolled and abnormal cell division leading to the proliferation of cells. The types of cancer are classified based on the origin of the organ or tissue and the molecular characteristics of the cancer cells [1]. After lung cancer, the most prevalent type of cancer worldwide is breast cancer [2]. Breast cancer can affect individuals of any age, both males and females, but it occurs more frequently in females over the age of 40. Each year, approximately 1 million cases are recorded globally, with 60% of these cases coming from low- and middle-income nations. In Pakistan, breast cancer is the leading cause of death in women due to cancer [3]. Several risk factors have been linked to breast cancer in women, including the presence of estrogen, postmenopause, late menopause, obesity, and high levels of endogenous estradiol [2,4-6]. Breast cancer is not uniformly caused at the molecular level; instead, it can be brought on by one or more factors. These molecular characteristics include activation of the HER2 (human epidermal growth factor receptor 2), activation of the oestrogen and progesterone receptors, and/or BRCA mutations [7].
The oncogene, Adipogenesis associated Mth938 domain containing (AAMDC), is believed to play a crucial role in the regulation of fat cell differentiation. According to NCBI, it is localized at 11q14.1 and functions in the cytoplasm [8]. In addition, AAMDC is known to work in conjunction with RNA polymerase II, positively regulating transcription and negatively regulating the apoptotic process. In situations of metabolic stress, such as estrogen deprivation, AAMDC is known to constitutively activate the PI3K-AKT-mTOR pathway, leading to the survival of ER+ breast cancers [9]. The PI3K/AKT/mTOR pathway is an important intracellular signaling mechanism that regulates the cell cycle, impacting cellular dormancy, proliferation, and cancer [10]. The AAMDC protein provides a protective shield to cancer cells, hindering their ability to be treated with anti-cancer hormone therapy. This protein has the ability to alter the metabolic processes of breast cancer cells, triggering growth pathways and facilitating their growth and division [11].
SNP refers to a variation in a single nucleotide at a specific position in the DNA sequence. It plays a crucial role in understanding the relationship between genetics and diseases [12]. In the human genome, SNPs account for over 90% of sequence variations and are used to identify genetic variations and biomarkers [13]. Owing to their widespread frequency, simplicity in analysis, affordability in genotyping, and the application of statistical and bioinformatics tools, single nucleotide polymorphisms (SNPs) are considered as the most useful biomarkers for the diagnosis of illnesses or prognosis [14]. The goal of SNP research in disease genetics is to identify single nucleotide polymorphisms (SNPs) that alter cellular biological processes and result in diseased states [15].
Materials and Methods
None of the SNPs study has specifically investigated the role of the AAMDC gene in the development of the Breast Cancer. This study aims to fill this gap by conducting a computational analysis of the AAMDC gene to identify any potential Single Nucleotide Polymorphisms (SNPs). The analysis is divided into two domains: sequence analysis of the gene for SNPs and structural and functional analysis of the gene. Both domains involve in-silico methods.
Data Retrieval and Pre-Processing
To gather information about the AAMDC gene, two well-known genomic databases, NCBI and ENSEMBL, were consulted. The data obtained from these sources included the gene's description, chromosomal location, transcripts, genomic segments, and resulting products. Additionally, non-synonymous single nucleotide polymorphisms (nsSNPs) obtained from these databases were analyzed to determine which nsSNPs were the most damaging. To achieve this, several bioinformatics tools were employed, including SIFT, PolyPhen-2, and CADD.
NCBI: NCBI houses numerous databases containing biological information, such as GenBank for DNA sequences and PubMed for biomedical literature [16].
ENSEMBL: Ensembl allows users to retrieve protein sequences, predict missense amino acids, perform multiple sequence alignments, annotate genes, and predict regulatory functions of proteins [17]. The SNPs of all the transcripts of AAMDC gene, including the missense Single Nucleotide Polymorphisms (SNPs), were extracted, processed manually in excel and documented.
Sequence Analysis
Sequence analysis was performed to check the damaging effect of the SNPs in our gene of interest i.e. AAMDC, using following tools.
Prediction of damaging SNPs: SNPs which have a negative impact on protein stability, mRNA, protein structure and function, are considered as "Damaging SNPs" and have been associated with various diseases. To determine the effect of SNPs on protein sequences, a comprehensive analysis was performed on a filtered set of most damaging non-synonymous SNPs using various bioinformatics tools.
SIFT: The SIFT (http://sift.bii.a-star.edu.sg) tool is a computational tool that predicts the functional impact of substitutions by taking into account the sequence homology and physical properties of amino acids, including naturally occurring non-synonymous polymorphisms and laboratory-induced missense mutations [18]. SIFT was used to analyze a set of most damaging 175 filtered non-synonymous single nucleotide polymorphisms (nsSNPs) from Ensembl data, with the aim of identifying nsSNPs that have a damaging effect on protein sequences.
POLYPHEN-2: Polyphen-2, Polymorphism Phenotyping v2, is a computational tool that assesses the impact of amino acid changes on the structure and function of human proteins. It utilizes basic physical and evolutionary principles to predict the functional effects of non-synonymous single nucleotide polymorphisms (nsSNPs) on a given protein [19]. The filtered set of most damaging 175 nsSNPs were added as input into Polyphen-2 to evaluate their potential for damaging effects on protein sequence and identify nsSNPs that have a detrimental impact on protein function.
CADD (Combined Annotation Dependent Depletion): The Combined Annotation Dependent Depletion (CADD) system calculates a unified score that takes into account multiple genomic annotations, by comparing the variations that have survived natural selection with those that are simulated mutations [20]. This system was used to analyze most damaging 175 shortlisted non-synonymous single nucleotide polymorphisms (nsSNPs) by incorporating them as input into the CADD system, to determine the potential deleterious effect of amino acid changes on the protein sequence.
Protein-Stability Prediction
Protein stability is the overall balance of forces that determines whether a protein will be in its natural, folded structure or a denatured (unfolded or stretched) condition. The prediction of protein stability was carried out by using following database.
I-Mutant 2: I-Mutant2.0 program utilizes Support Vector Machine (SVM) algorithms to predict the effect of single point mutations on protein stability. as quantified by DeltaDeltaG values, and classifies the direction of the stability change resulting from the mutation [21]. To evaluate the impact of amino acid changes on protein structure, most damaging 175 nsSNPs with both new and wild type sequences were added as input into the I-Mutant2.0 program
Disease Association Prediction
The process of Disease Association Prediction aims to assess the detrimental effects of nsSNPs on the specified gene using bioinformatics tools.
PhD SNP: PhD SNP (https://snps.biofold.org/phd-snp/phd-snp.html) operates using the FASTA format for protein sequences and was designed to assess the damaging effects of amino acid changes in protein structure and their correlation with various diseases associated with the AAMDC gene [22]. To accomplish this, most damaging 175 nsSNPs, consisting of both new and wild type variations, were incorporated as input into the PhD SNP for analysis.
PANTHER: Panther (Protein Analysis Through Evolutionary Relationships) tool is designed to evaluate the potential impact of nsSNPs on protein function. The score for a given nsSNP is determined through a Hidden Markov Model (HMM) alignment of evolutionary related proteins [23].
Structural and Functional Prediction
The structural and functional changes in the protein due to nsSNP were assessed using the bioinformatics tool given below
MUTPRED2: MutPred2 is a tool that categorizes nsSNPs as either harmful or benign, and predicts their effect on over 50 different protein characteristics [24]. MutPred2 was used to evaluate the structural and functional effects of most damaging 175 nsSNPs.
SNAP2: SNAP2 is a bioinformatics tool that utilizes neural networks to predict the impact of single amino acid variations on a protein's function [25].
Results and Discussion
Data Retrieval and Pre-Processing
The National Center for Biotechnology Information (NCBI) is a highly regarded repository for biotechnology and biomedicine-related resources and databases. The gene known as AAMDC has been assigned alternate designations, including CK067, PTD015, and C11orf67. This gene is located on the 11q14.1 chromosomal region and consists of 12 exonic regions [26].
The Ensembl genome browser was used to retrieve the transcripts and corresponding sequences of the AAMDC gene. The gene has 11 transcript variants and 207 orthologues, as depicted in Table 1. The SNPs of all transcripts were documented processed manually in excel and nsSNPs were shortlisted in excel.
![]()
Transcript ID
Name
Amino Acids
Biotype
ENST00000393427.7
AAMDC-202
122aa
Protein Coding
ENST00000527134.5
AAMDC-207
137aa
Protein Coding
ENST00000532481.5
AAMDC-210
88aa
Protein Coding
ENST00000304716.12
AAMDC-201
147aa
Protein Coding
ENST00000533193.5
AAMDC-211
168aa
Protein Coding
ENST00000526415.5
AAMDC-206
122aa
Protein Coding
ENST00000525034.1
AAMDC-203
122aa
Protein Coding
ENST00000525409.5
AAMDC-204
90aa
Protein Coding
ENST00000526164.5
AAMDC-205
93aa
Nonsense Mediated Decay
ENST00000529666.1
AAMDC-208
51aa
Nonsense Mediated Decay
ENST00000531855.1
AAMDC-209
No protein
Protein Coding CDS not defined
Table 1: Ensembl genome browser showing Transcripts of AAMDC Gene.
Sequence Analysis
Prediction of damaging SNPs: Prediction of damaging SNPs was done by using various state-of-the art algorithms (SIFT, POLYPHEN-2, and CADD). SIFT version 2.0 predicts an amino acid substitution score from zero to one. Out of most damaging 175 nsSNPs, 160 nsSNPs were labeled as ‘deleterious’ whereas 15 were labeled as ‘Tolerated’. Out of most damaging 175, 160 were predicted as ‘Probably Damaging’, 13 as ‘Possibly Damaging’ whereas only one SNP was predicted as ‘Benign’ by POLYPHEN-2. CADD predicted 38 nsSNPs as ‘Deleterious’ whereas 137 were predicted as ‘Benign’. All the most damaging 175 nsSNPs were then shortlisted on consensus base to check for the nsSNPs which damaging in all the tools. 10 nsSNPs were predicted to be damaging SNPs as highlighted in table 2.
![]()
Mutations
Transcripts
SIFT
Polyphen-2
CADD
I6T
ENST00000393427.7
ENST00000527134.5
ENST00000532481.5
ENST00000304716.12
ENST00000533193.5
ENST00000526415.5
ENST00000525034.1
ENST00000525409.5
ENST00000526164.5
ENST00000529666.1
ENST00000531855.1Deleterious
Probably Damaging
Likely Deleterious
W11G
Deleterious
Probably Damaging
Likely Deleterious
D25N
Deleterious
Probably Damaging
Likely Deleterious
P30S
Deleterious
Probably Damaging
Likely Deleterious
G31A
Deleterious
Probably Damaging
Likely Deleterious
V49A
Deleterious
Probably Damaging
Likely Deleterious
V66M
Deleterious
Probably Damaging
Likely Deleterious
A68T
Deleterious
Probably Damaging
Likely Deleterious
L75S
ENST00000532481.5
&
ENST00000533193.5Deleterious
Probably Damaging
Likely Deleterious
A146T
ENST00000533193.5
Deleterious
Probably Damaging
Likely Deleterious
Table 2: Consensus based predicted list of damaging SNPs.
Protein-Stability Prediction
I-Mutant2.0 predicts protein stability changes upon single nucleotide polymorphism. All the shortlisted most damaging 175 nsSNPs with mutant and wild type were added to the I-Mutant2.0 as an input to check damaging effect of amino acid changes in protein structure. Out of most damaging 175, 136 nsSNPs predicted the ‘Decrease’ in protein stability whereas 39 nsSNPs predicted ‘Increase’ in protein stability. Ten census based shortlisted nsSNPs were also added in I-Mutant2.0. I-Mutant2.0 predicted that there was a large decrease in stability of protein for 10 SNPs variant persisted as shown in the table 3, which highlighted that mutations affected the stability of structure of protein. Reliability Index value of I-Mutant2.0 is also given in the table 3. Based on the result of I-Mutant2.0, a consensus based predicted list of protein stability is highlighted in the table 3.
![]()
Mutations
Transcripts
I-Mutant2.0
Reliability Index
I6T
ENST00000393427.7
ENST00000527134.5
ENST00000532481.5
ENST00000304716.12
ENST00000533193.5
ENST00000526415.5
ENST00000525034.1
ENST00000525409.5
ENST00000526164.5
ENST00000529666.1
ENST00000531855.1Decrease
8
W11G
Decrease
10
D25N
Decrease
8
P30S
Decrease
8
G31A
Decrease
8
V49A
Decrease
9
V66M
Decrease
9
A68T
Decrease
9
L75S
ENST00000532481.5
&
ENST00000533193.5Decrease
9
A146T
ENST00000533193.5
Decrease
9
Table 3: Consensus based predicted list of Protein Stability due to nsSNPs.
Disease Association Prediction
All the shortlisted most damaging 175 nsSNPs with mutant and wild type were added to the PhD SNP as an input to check damaging effect of amino acid changes in protein structure and their association with different diseases associated with AAMDC gene. Out of most damaging 175, 94 were predicted as ‘Disease’ whereas 86 were ‘Neutral’ and reliability index ranged from 0-10. The results of PANTHER indicated that 156 nsSNPs were predicted as "probably damaging," while 19 were deemed "benign". Based on the results of PhD-SNP and PANTHER, a consensus based predicted list of Disease-Associated SNPs overlapped with previous consensus is given in the table 4.
![]()
Mutations
Transcripts
PhD SNP
PANTHER
W11G
ENST00000393427.7
ENST00000527134.5
ENST00000532481.5
ENST00000304716.12
ENST00000533193.5
ENST00000526415.5
ENST00000525034.1
ENST00000525409.5
ENST00000526164.5
ENST00000529666.1
ENST00000531855.1Disease
Probably Damaging
P30S
Disease
Probably Damaging
G31A
Disease
Probably Damaging
Table 4: Consensus based predicted list of Disease-Associated SNPs.
Structural and Functional Prediction
All the most damaging 175 nsSNPs were subjected to MutPred2 to analyze probability of deleterious mutation score. The results showed that some nsSNPs variants highlighted Molecular mechanisms such as gain of instrinsic disorder, altered ordered interface, Loss of Relative solvent accessibility, Altered Stability, Loss of N-linked glycosylation, Altered Metal binding, Loss of Loop, Gain of Disulfide linkage and altered trans membrane protein whereas other highlighted the affected prosite as well as different ELM Motifs e.g. ELME000052 & ELME000335. The result included predicted molecular mechanism, property score (Pr) of molecular mechanism and its P-value, Predicted conservation scores, affected prosite and ELM Motifs. All the most damaging 175 variants were also added as an input in SNAP2 tool. Out of damaging 175 nsSNPs, 120 nsSNPs showed ‘Effect’ on structural phenotype of proteins and 55 were ‘Neutral’. Based on the results of MUTPRED2 and SNAP2, a consensus based predicted list of structural analysis of nsSNPs is given in the table 5.
![]()
Mutations
Transcripts
MUTPRED2
SNAP2
W11G
ENST00000393427.7
ENST00000527134.5
ENST00000532481.5
ENST00000304716.12
ENST00000533193.5
ENST00000526415.5
ENST00000525034.1
ENST00000525409.5
ENST00000526164.5
ENST00000529666.1
ENST00000531855.10.888
Effect
85%P30S
0.768
Effect
63%G31A
0.666
Effect
66%
Table 5: Consensus based predicted list of Structural Analysis by bioinformatics tools.
Consensus Based Deleterious nsSNPs
Three nsSNPs common in transcripts ENST00000393427.7, ENST00000527134.5, ENST00000532481.5, ENST00000304716.12, ENST00000533193.5, ENST00000526415.5, ENST00000525034.1, ENST00000525409.5, ENST00000526164.5, ENST00000529666.1 and ENST00000531855.1 were identified as most deleterious nsSNPs after analysis of most damaging 175 nsSNPs in AAMDC gene, as shown in figure 2 and table 6. According to consensus based table 6, the deleterious nsSNPs variants of the AAMDC gene were highly predicted as damaging based on consensus approach using the results retrieved from sequence analysis, prediction of disease association, structural analysis and functional analysis.
![]()
Bioinformatics Tools
W11G
P30S
G31A
SIFT
Damaging
Damaging
Damaging
Polyphen-2
Probably Damaging
Probably Damaging
Probably Damaging
CADD
Deleterious
Deleterious
Deleterious
I-Mutant2.0
Decrease
Decrease
Decrease
PhD-SNP
Disease
Disease
Disease
PANTHER
Probably Damaging
Probably Damaging
Probably Damaging
SNAP2
Effects
Effects
Effects
Mutpred-2
0.888
0.768
0.666
Table 6: Consensus based result of all tools for deleterious nsSNPs in AAMDC Gene.
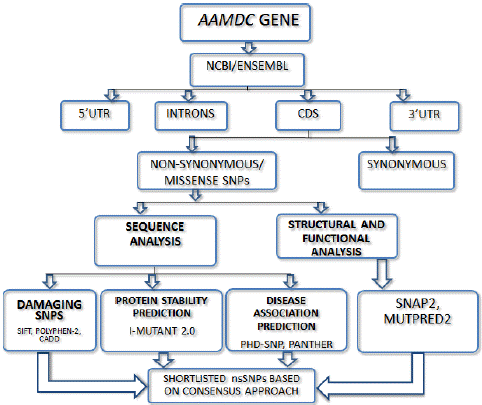
Figure 1: Graphical representation of the Methodology.
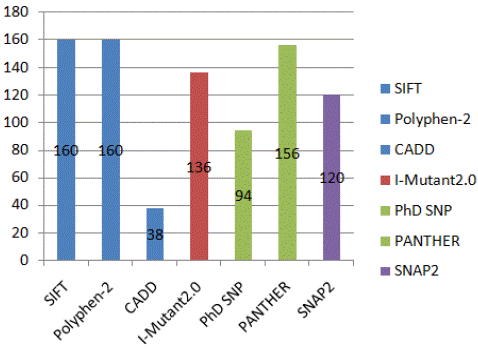
Figure 2: Consensus based graphical view nsSNPs using bioinformatics tools to determine sequence analysis, protein stability, disease association, structural and functional prediction.
Structural Analysis
Structural analysis was performed to determine the structural changes between the wild and mutant deleterious SNPs that were predicted by the bioinformatics tools through sequence analysis of AAMDC gene. Structural Analysis was conducted in two stages.
HOPE: The HOPE (Hyperactive Optimized Protein Engineering) methodology was utilized to evaluate the impact of mutagenesis on the wild and mutant forms of a gene. The analysis revealed disparities in various aspects, such as the amino acid composition, structural features, domains, physical characteristics, hydrophobicity, and spatial arrangement, between the native and altered amino acid residues. HOPE was applied to three consensus-based, deleterious nsSNPs in the AAMDC gene, specifically W11G, P30S, and G31A, to compare the wild and mutated protein sequences.
W11G variant: The W11G variant of the AAMDC gene has been predicted to result in a structural change through a mutation as identified by the HOPE algorithm. This mutation involves a substitution of Tryptophan with Glycine at position 11 in the protein's structure. The mutant residue, as depicted in figures 3, 4 and 5, has a smaller size and lower hydrophobicity compared to the wild-type residue. The protein ribbon structure is illustrated in figure 4, with a close-up view of the mutation presented in figure 5. The side chains of the protein, mutant and wild residue are respectively depicted in gray, red, and green.
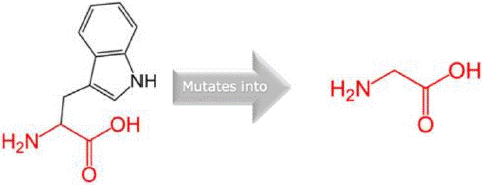
Figure 3: The structural change in protein due to mutation from W to G at 11 in AAMDC protein as predicted by HOPE.
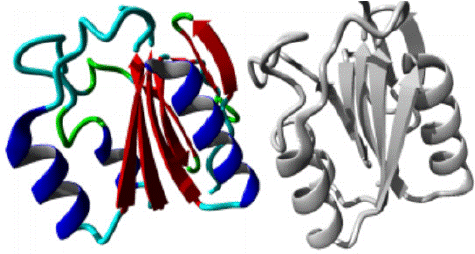
Figure 4: AAMDC protein in ribbon structure. a-helix, β-strand, turn, other molecules in the complex are colored blue, red, green and grey respectively, as predicted by HOPE.
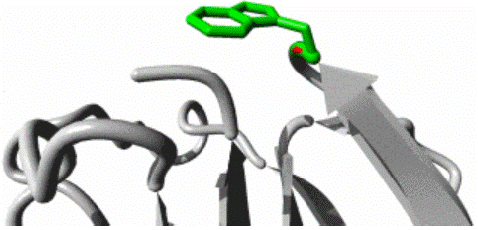
Figure 5: Close up view of wild and mutant residue in AAMDC protein as predicted by HOPE.
P30S variant: The HOPE analysis predicted that a structural alteration from Proline to Serine at position 30 in the protein structure would occur, as depicted in Figure 6. The amino acids are depicted in black and red, representing the side chains and functional groups, respectively. According to HOPE, the wild residue Proline is highly rigid, and thus, its mutation could disrupt the unique conformational structure of the protein. Additionally, the analysis indicated that this residue is located within the core of the Mth938 domain and, therefore, the mutation could also alter or compromise the core structure of the Mth938 domain. The protein ribbon structure is illustrated in figure 7. A close-up view of the mutation is shown in figure 8, with the protein, mutant, and wild residue side chains depicted in gray, red, and green, respectively.

Figure 6: The structural change in protein due to mutation from P to S at 30 in AAMDC protein as predicted by HOPE.
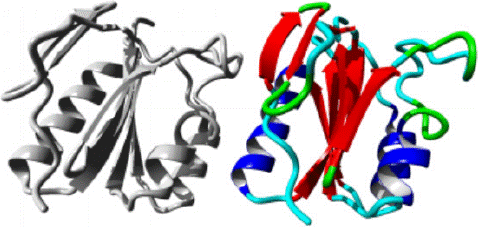
Figure 7: AAMDC protein in ribbon structure. a-helix, β-strand, turn, other molecules in the complex are colored blue, red, green and grey respectively, as predicted by HOPE.
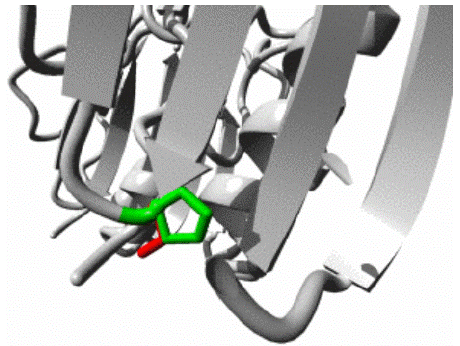
Figure 8: Close up view of wild and mutant residue in AAMDC protein as predicted by HOPE.
G31A variant: HOPE predicted a structural change from Guanine to Alanine occurs at position 31 in protein structure as shown in figure 9. Backbone in the figure is colored red whereas side chain is colored black. HOPE predicted that mutant amino acid is bigger in size than wild amino acid. The mutant amino acid is more hydrophobic than wild. The wild type residue Glycine is the most flexible of all the amino acids, the mutation in which will result in loss of this function. The protein ribbon structure is shown in figure10. The close up mutation is shown in figure 11, along with protein, mutant and wild residue side chain in grey, red and green respectively.

Figure 9: The structural change in protein due to mutation from G to A at 31 in AAMDC protein as predicted by HOPE.
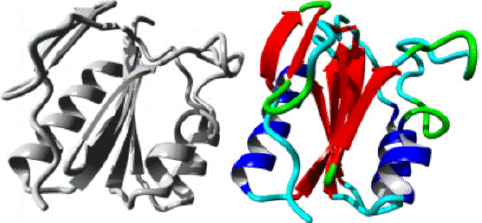
Figure 10: AAMDC protein in ribbon structure. a-helix, β-strand, turn, other molecules in the complex are colored blue, red, green and grey respectively, as predicted by HOPE.
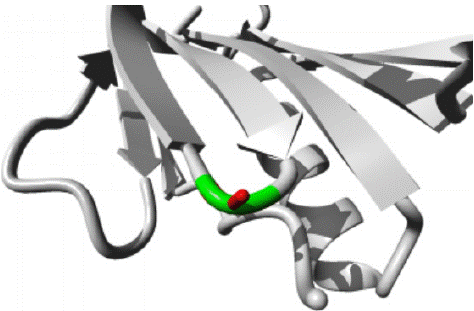
Figure 11: Close up view of wild and mutant residue in AAMDC protein as predicted by HOPE.
STRING:
The molecular docking predictions performed using STRING software have revealed the potential interactions between the AAMDC protein and ten other proteins, including KCTD14 (a BTB/POZ domain-containing protein involved in potassium channel tetramerization), GATSL3 (a cytosolic arginine sensor for the mTORC1 signaling pathway), AAED1 (a thioredoxin-like antioxidant enzyme), INTS4 (a component of the Integrator complex involved in small nuclear RNA transcription and processing), AADACL3 (an arylacetamide deacetylase-like protein), RNPEP (an arginyl aminopeptidase), AASDH (an aminoadipate-semialdehyde dehydrogenase), NARS2 (a probable mitochondrial asparagine-tRNA ligase), AAR2 (a cistron-splicing factor in volved in pre-mRNA splicing), and AADACL4 (an arylacetamide deacetylase-like protein) as shown in figure 12. These interactions are crucial for understanding the biological processes involving these proteins.
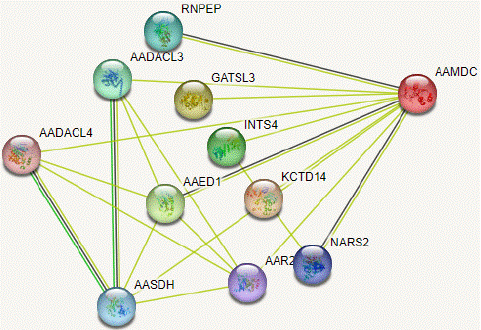
Figure 12: Protein-Protein interaction network of AAMDC gene using STRING server.
Conclusion
In this study, the impact of deleterious single nucleotide polymorphisms (nsSNPs) on the coding regions of the AAMDC gene was evaluated using bioinformatics tools. This is the first research to predict the impact of non-synonymous SNPs (nsSNPs) on the structure and function of the AAMDC protein. Out of most damaging 175 missense SNPs, three SNPs (W11G, P30S, and G31A) were predicted to be the most deleterious. These nsSNPs were found to have a negative impact on the protein's structure and function. The W11G and G31A nsSNPs have a high probability of affecting transmembrane protein function, while the P30S nsSNP not only affects transmembrane protein but also impairs metal binding and activation of the active site. Structural analysis using the HOPE and STRING methods confirmed the impact of these deleterious nsSNPs. Individuals with these nsSNPs in their genome are more susceptible to mutations in AAMDC gene and therefore, an increased risk of breast cancer. This in-silico study can potentially aid in the development of effective drugs against Breast Cancer.
References
- Krieghoff-Henning E, Folkerts J, Penzkofer A, Weg-Remers S. Cancer – an overview. Med Monatsschr Pharm. 2017; 40: 48-54.
- Fakhri N, Chad MA, Lahkim M, Houari A, Dehbi H, Belmouden A, et al. Risk factors for breast cancer in women: an update review. Med Oncol. 2022; 39: 197.
- Ullah Z, Khan MN, Din ZU, Afaq S. Breast cancer awareness and associated factors amongst women in Peshawar, Pakistan: A cross-sectional study. Breast Cancer (Auckl). 2021; 15: 11782234211025346.
- Kashyap D, Pal D, Sharma R, Garg VK, Goel N, Koundal D, et al. Global increase in breast cancer incidence: risk factors and preventive measures. BioMed Res Int. 2022; 2022: 9605439.
- Key TJ, Verkasalo PK, Banks E. Epidemiology of breast cancer. Lancet Oncol. 2001; 2: 133-40.
- Arif S, Baloch Q, Zaheer F, Agheem R, Ariff M, Ahmed M. The adequate breast cancer knowledge assessment: A cross-sectional study done among nonmedical women of Karachi. J Educ Health Promot. 2018; 7: 169.
- Zhang G, Ren C, Li C, Wang Y, Chen B, Wen L, et al. Distinct clinical and somatic mutational features of breast tumors with high-, low-, or non-expressing human epidermal growth factor receptor 2 status. BMC Med. 2022; 20: 142.
- NCBI. AAMDC adipogenesis associated Mth938 domain containing [Homo sapiens (human)]. 2022.
- Golden E, Rashwan R, Woodward EA, Sgro A, Wang E, Sorolla A, et al. The oncogene AAMDC links PI3K-AKT-mTOR signaling with metabolic reprograming in estrogen receptor-positive breast cancer. Nat Commun. 2021; 12: 1920.
- King D, Yeomanson D, Bryant HE. PI3King the lock: targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J Pediatr Hematol Oncol. 2015; 37: 245-51.
- Perkins H. Gene found for deadliest breast cancer; 2021.
- Puthiyedth N, RN. Feature selection methods for SNP analysis. In: 2nd International Conference on Intelligent Computing, Instrumentation and Control Technologies (ICICICT). 2019; 2019.
- Chang K, Deng S, Chen M. Novel biosensing methodologies for improving the detection of single nucleotide polymorphism. Biosens Bioelectron. 2015; 66: 297-307.
- Srinivasan S, Clements JA, Batra J. Single nucleotide polymorphisms in clinics: fantasy or reality for cancer? Crit Rev Clin Lab Sci. 2016; 53: 29-39.
- Schork NJ, Fallin D, Lanchbury JS. Single nucleotide polymorphisms and the future of genetic epidemiology. Clin Genet. 2000; 58: 250-64.
- Sayers EW, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021; 49: D10-d17.
- Newman V, Moore B, Sparrow H, Perry E. The Ensembl genome browser: strategies for accessing eukaryotic genome data. Methods Mol Biol. 2018; 1757: 115-39.
- Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016; 11: 1-9.
- Frigeni M, Balakrishnan B, Yin X, Calderon FRO, Mao R, Pasquali M, et al. Functional and molecular studies in primary carnitine deficiency. Hum Mutat. 2017; 38: 1684-99.
- LeDoux MS. Population prevalence of deleterious SGCE variants. Tremor Other Hyperkinet Mov (N Y). 2020; 10: 50.
- Behrendt A, Golchin P, König F, Mulnaes D, Stalke A, Dröge C, et al. Vasor: accurate prediction of variant effects for amino acid substitutions in multidrug resistance protein 3. Hepatol Commun. 2022; 6: 3098-111.
- Irfan M, Iqbal T, Hashmi S, Ghani U, Bhatti A. Insilico prediction and functional analysis of nonsynonymous SNPs in human CTLA4 gene. Sci Rep. 2022; 12: 20441.
- Mi H, Ebert D, Muruganujan A, Mills C, Albou LP, Mushayamaha T, et al. PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021; 49: D394-403-d403.
- Havranek B, Islam SM. Prediction and evaluation of deleterious and disease causing non-synonymous SNPs (nsSNPs) in human NF2 gene responsible for neurofibromatosis type 2 (NF2). J Biomol Struct Dyn. 2021; 39: 7044-55.
- Nawar N, Paul A, Mahmood HN, Faisal MI, Hosen MI, Shekhar HU. Structure analysis of deleterious nsSNPs in human PALB2 protein for functional inference. Bioinformation. 2021; 17: 424-38.
- NCBI. AAMDC adipogenesis associated Mth938 domain containing. 2022.