
Research Article
Austin J Clin Neurol. 2025; 12(1): 1170.
Role of Neuregulin1 in Tumor Necrosis Alpha Induced Blood Brain Barrier Injury
Stewart W, Hejl C+, Guleria RS* and Gupta S*
Translational Science Core, VISN 17 Center of Excellence for Research on Returning War Veterans, Central Texas Veterans Health Care System, 4800 Memorial Drive (151C), Waco, Texas, 76711, USA +Equally Contributed to the Manuscript
*Corresponding author: Sudhiranjan Gupta, Translational Science Core, VISN 17 Center of Excellence for Research on Returning War Veterans, Central Texas Veterans Health Care System, 4800 Memorial Drive (151C), Waco, Texas, 76711, USA Email: Sudhiranjan.Gupta@va.gov
Rakeshwar S Guleria, Translational Science Core, VISN 17 Center of Excellence for Research on Returning War Veterans, Central Texas Veterans Health Care System, 4800 Memorial Drive (151C), Waco, Texas, 76711, USA Email: Rakeshwar.Guleria@va.gov
Received: February 11, 2025; Accepted: March 03, 2025; Published: March 06, 2025
Abstract
Neuroinflammation is a key feature after traumatic brain injury (TBI) in the central nervous system (CNS), characterized by increased pro- and antiinflammatory cytokines, disruption of blood brain barrier (BBB) permeability, resulted in many different neurological diseases. Furthermore, perturbation of BBB is pivotal in TBI as dysfunctional barrier allows inflammatory and immune cells trafficking leading to the impairment of the neurovascular unit. Although several drugs have been shown to exhibit promising effects for TBI, the restoration of BBB damage remain elusive. Neuregulin 1 (Nrg1) is a growth factor plays multiple functions in CNS, however, it’s role in inflammation-induced BBB damage is unknown. We hypothesize that Nrg1 may protect inflammationinduced BBB impairment. In the current study, we investigated the protective effects of Nrg1 in Tnfa-stimulated gene expression of several tight junction proteins, inflammatory genes, NF-kB activation, apoptosis, in human brain microvascular endothelial cells (hBMVECs), one of the critical cell types in the BBB integrity. These data suggest that pretreatment with Nrg1 reversed the Tnfa-induced damage of BBB components in hBMVECs. Furthermore, these results identified neuroplilin1 as a possible target for Nrg1. Together, data provided evidence that Nrg1 can serve as an anti-inflammatory molecule and may offer a promising therapeutic target in BBB damage. In conclusion, we propose that Nrg1-NF-kB-axis in BBB impairment is vital in TBI pathophysiology and warrant further investigation.
Keywords: Blood Brain Barrier; Inflammation; NF-kB; Neuregulin1
Introduction
The blood brain barrier (BBB) serves as a selective semipermeable sheath separating the central nervous system (CNS) from systemic circulation. The sheath is a bi-directional system which controls the traffic of solutes, nutrients and toxins via efflux transporters and maintains the brain homeostasis [1]. Human BBB vasculature is composed of brain microvascular endothelial cell (BMVEC) monolayer along with other neuronal cells like pericytes, astrocytes, glial cells, forming coherent interconnectivity at the barrier [2]. The BMVEC is held together by tight junctions (TJ) which are composed of Occludin, Claudins, zonula Occludens (ZO-1, ZO-2, ZO-3); etc.; adherent junctions (AJ) composed of cadherins, catenins, vinculin, and actinin; and junctional adhesion molecules (JAM) [3,4]. Together these cells regulate cellular transport properties. The BMVEC coupled with astrocytes, pericytes, neurons and extracellular matrix constitute the neurovascular unit (NVU) and contributes BBB functional integrity [5]. Inflammation, a critical contributor in traumatic brain injury (TBI) impaired BBB integrity and elicits both short- and long-term effects. Neuroinflammation is further suggested as secondary injury to many neurological diseases including TBI [6,7]. After an episode of TBI, BBB injury results in alteration of the microenvironment and pathological sequels in the NVU like edema, apoptosis and neuroinflammation [7-10]. Recent reports suggested that perturbation of the BBB is crucial in TBI as impairment of the barrier allows inflammatory and immune cell trafficking leading to the development of a neuroinflammatory atmosphere in the NVU [11-17].
In our study we have chosen human BMVEC (hBMVECs) as an in vitro model system to understand the underlying molecular mechanism of inflammation induced BBB injury. We used Tumor Necrosis Factor alpha (TNF-a) as a proinflammatory cytokine agent that triggers BBB dysfunction [18-20].
Neuregulin 1 (Nrg1) belongs to the family of growth factors that contain epidermal growth factor-like domain, bind to ErbB receptors, dimerize, and activate downstream signaling cascade [21,22]. The endogenous ligand for Nrg1 is ErbB family which consisting of four ligands, ErbB1-4 [22]. ErbB2 does not bind to Nrg1 but has strong kinase activity. At the receptor level, only ErbB3 and ErbB4 can bind to neuregulin (Nrg1-3) [23]. Nrg1 has also shown diverse function in neuron and glia and, works mainly through the activation of ErbB 4 receptor tyrosine kinases [24]. Nrg1 has shown potential in neuroprotection in ischemia-induced neuronal death in rodent model [24-30]. Neuroinflammation is a key process in BBB damage and Nrg1 has shown potential as an anti-inflammatory molecule in microglial cells and, also in ischemic stroke model [27,28]. However, role of Nrg1 in inflammation-induced BBB damage is currently unknown. We hypothesize that Nrg1 may protect inflammation-induced BBB in hBMVECs.
The present study is designed to investigate the changes of TJ protein because these proteins are responsible for free solute exchange between the CNS and vasculature during injury. We report that TNFa-induced inflammation in hBMVECs is associated with downregulation of TJ genes and proteins, upregulation of several inflammatory genes, activation of Nuclear Factor kappa B (NFkB); lead to enhance the cellular permeability that resulted in BBB dysfunction. All these above-mentioned alterations were restored following the pretreatment with recombinant Nrg1 protein (rNrg1). Altogether, these data provided evidence that Nrg1 can serve as an anti-inflammatory molecule and may offer a promising therapeutic target in BBB damage.
Materials and Methods
Cell Culture and Treatment
Primary hBMVECs (passage 2, cat no. ACBRI 376) were purchased from Cell Systems, Kirkland, WA, USA and cultured as described previously [31]. In brief, cells were allowed 48 h for growth to a field density of 2.5×105 cells per cm2 before serum starvation (2h) with complete serum free medium. The cells were then pretreated with Nrg1 human recombinant protein (rNrg1, cat. No Ab50227, Abcam, USA) at a concentration of 100 ng/mL for 2 hours before Tnfa (cat no. ab9642, Abcam) stimulation. The stimulation dose for of Tnfa was 25 ng/mL (Sigma Aldrich, USA) which did not show any toxic effect or damage to the cells and was used throughout the study. The current work was carried under an approved research protocol which was reviewed and approved by the Central Texas Veterans Health Care System (CTVHCS), Research and Development Committee.
RNA Isolation and Quantitative Real-Time PCR (Q-RT-PCR) Analyses
Total RNAs from the hBMVECs were extracted using RNeasy kit (Qiagen, Valencia, CA) as per the manufacturer's instructions. For real-time RT-PCR, 200 ng of total RNAs were reverse transcribed to cDNA using cDNA synthesis kit (OriGene, Rockville, MD, USA) by following the manufacturer's instructions. Quantitative PCR was performed as described previously [31-33]. Analysis of gene expression was evaluated by 2(ΔΔCt) method. Each reaction was in triplicate, repeated three times and GAPDH was used as an internal loading control. The gene-specific primers used for the study were purchased from OriGene Technologies, Inc., USA.
Immunofluorescence Microscopy
The hBMVECs were seeded in chamber slides and were treated as described previously [31]. In brief, cells were washed with 1X PBS followed by cell fixation with paraformaldehyde and permeabilization, cells were blocked in 5% blocking buffer for 60 min at room temperature and incubated with anti‐ZO3 (D57G7 XP), anti-Occludin (E6B4R) and anti-NF-kB-p65 primary antibodies (1:1000) separately overnight at 4°C in antibody dilution buffer (1% blocker BSA in PBS and 0.01 % Triton X-100). The cells were then washed (three times with 1X PBS), incubated with corresponding secondary antibodies (1:1,000) for 1-hour. A cover glass was mounted over the growth area with a drop of ProLong Diamond antifade mounting media containing 4, 6-daimindo-2-phenylindole (DAPI). Fluorescence images at 20X magnification were captured by Leica DMi8 Imaging System. All the primary and the secondary antibodies were purchased from Cell Signaling Technology, Beverly, MA, USA. Anti-ZO3 (cat. no. 3704), anti-Occludin (cat. no. 91131), anti-p65 (cat. no. 8242); were purchased from Cell Signaling Technology, Inc., USA. ProLong Diamond antifade mounting solution containing DAPI (cat. no. P36962) was purchased from Thermo Fischer Scientific, Inc., USA. Cells were counted in 10-15 randomly chosen microscopic fields (average of 80-100 cells per treatment) and compared with the experimental and control groups and are representative of three separate experiments.
Western Blotting
Cell lysate preparation and Western blot experiments were performed as we previously described [29,32]. In total, 40 μg cell lysates were used for Caludin5, 15 μg cell lysates were used for Occludin, and 20 μg cell lysate was used for Cleaved Caspase 3 analyses, respectively. The images were taken in every step using ChemiDoc MP imaging system (BioRad, USA). Primary antibodies against Occludin (cat. no. 91131), Claudin5 (cat no 495645), Cleaved Caspase3 (cat. no. 9664) and NF-kB-p65 (cat no 8242) were purchased from Cell Signaling Technology, Inc., USA. The immunoreactive bands were visualized using Clarity Max Western enhanced chemiluminescence (ECL) kit (cat. no. 1705062, Bio‐Rad Laboratories, Inc., USA), and later the density of the band was quantified and analyzed using the ImageJ 4.1 software (National Institutes of Health). We ran the parallel blot for GAPDH control with the same amount of protein as ran for the target proteins.
Permeability and Trans Endothelial Electrical Resistance (TEER) Assay
The permeability was performed using Endothelial Trans-well Permeability Assay Kit (cat no. CB6929 from Cell Biologics, Inc., USA) according to manufacturer’s instruction. Data expressed as A450 absorption readings are considered a relative permeability. The BBB function was determined by TEER assay using Milicell ERS2 (Milipore, Massachusetts, USA) volt-ohm meter according to the manufacturer’s protocol. In brief, the hBMVEC, 1X 105 cells were seeded in gelatin-coated permeable polyester Transwell filter inserts (Corning Coster, Thermo Fischer Scientific, Inc. USA). The treatment for Tnfa and rNrg1 was described above in cell culture and treatment section. The TEER values were obtained by immersing the electrode so that the shorter tip is in the Millicell® culture plate inserts, and the longer tip is in the outer well. The volt-ohm meter captured the transmembrane electrical resistance according to the current and was measured. A blank transwell without cells but the medium was used as a blank control. All data was collected and analyzed from three independent experiments were performed in triplicate.
Statistical Analyses
All data are presented as means ± Standard Error Mean (SEM) from three independent experiments. For data analysis, unpaired t test or one-way analysis of variance (ANOVA) were performed using Prism 5.0 Graph Pad software (Graph Pad, San Diego, CA, USA). A P-value less than 0.05 was considered statistically significant.
Results
Effects of rNrg1 on Tnfa-stimulated Alteration of TJ mRNA and Protein in hBMVECs
To determine the effects of Tnfa on the TJ gene expression, hBMVECs were stimulated with 25 ng/ml Tnfa for 24 h. Tnfa treatment significantly decreased mRNA expression of tight junction protein 1(Tjp1), Tjp3, and Claudin 3 (P<0.05), compared with untreated control cells (Figure 1A, 1C and 1D). Our data did not show any change for Tjp2 mRNA expression (Figure 1B). Pretreatment with rNrg1 significantly ward off the reduction in their expression (P<0.05; Figure 1A, 1C and 1D). Together, our data suggested that rNrg1 pre-treatment protected Tnfa -induced downregulation of TJ gene expression in hBMVEC.
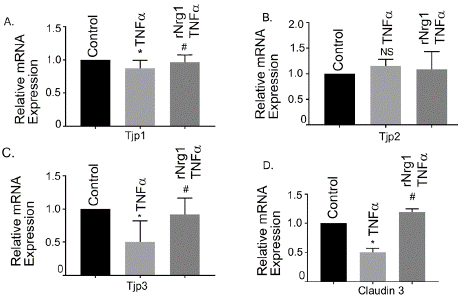
Figure 1: Effect of rNrg1 in Tnfa-induced tight junction gene expression in
human brain microvascular endothelial cells. hBMVECs were pretreated
with rNrg1 for 2h and stimulated with Tnfa at 25 ng/ml for a period of 24h.
The mRNA expression of (A) Tjp11, (B) Tjp2, (C) Tjp3 and (D) Claudin 3
were measured using reverse transcription-quantitative PCR. Results are
expressed as the mean ± SEM from 3 independent experiments. *P<0.05 vs.
untreated cells. NS, non-significant.
Furthermore, our data showed significant reduction of two critical TJ genes, Occludin and Claudin 5 along with their protein level as well (Figure 2A and 2B). The western blot analyses showed that protein levels of both Occludin and Claudin 3 significantly reduced after Tnfa stimulation, compared to control or unstimulated cells (Figure 2C). Pretreatment of rNrg1 significantly prevented the reduction of both the proteins.
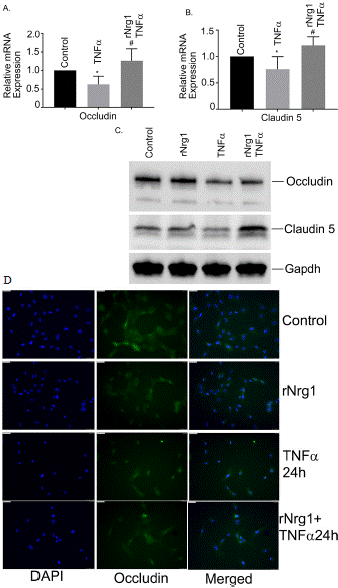
Figure 2: Effect of rNrg1 in Tnfa-induced Occludin and Claudin 5 levels in
human brain microvascular endothelial cells. hBMVECs were pretreated
with rNrg1 for 2h and exposed to Tnfa at 25 ng/ml for a period of 24h.
The mRNA expression of (A) Occludin and (B) Claudin 5 were measured
using reverse transcription-quantitative PCR. (C) Representative image of
the Western blot analysis of Occludin protein and Claudin 5 level. These
experiments were replicated three-to-five times, and amplifications were
performed and normalized to GAPDH. (D) Cultured hBMVECs were pretreated
with rNrg1 for 2h and then stimulated with Tnfa for 24h. Dual staining
immunofluorescence analysis of Occludin is shown in green. Blue staining
was DAPI, for the nucleus.
Finally, under similar conditions we evaluated Occludin levels by immunofluorescence staining. We observed that Tnfa treatment significantly reduced the cellular Occludin protein levels as shown little-to-no green fluorescence outside the nucleus, Figure 2D. However, TNFa induced Occludin degradation was restored with pretreatment of rNrg1 as evidenced by green fluorescence. Together, our data indicated that rNrg1 pre-treatment restored Tnfa-stimulated Occludin reduction in hBMVECs.
Effects of rNrg1 in Tnfa-stimulated Inflammatory Response Observed in hBMVECs
Inflammation plays a crucial role in BBB injury. We evaluated a set of inflammatory genes; the interleukin-6 (Il6), interleukin-1 b (Il1b) and C-C chemokine ligand 2 (CCL2) and NLR family pyrin domain containing protein 3 (NLRP3, an inflammasome), in Tnfa stimulated hBMVEC. Tnfa treatment showed a marked enhancement of the mRNA expression of Il1b, Il6, CCL2 and NLRP3 compared with their unstimulated cells (Figure 3 A-D). However, cells pre-treated with rNrg1, significantly showed reduction in the inflammatory response (Figure 3 A-D). Together, our data indicated that rNrg1 pre-treatment attenuated Tnfa-induced inflammatory response in hBMVECs and may serve as an anti-inflammatory molecule.
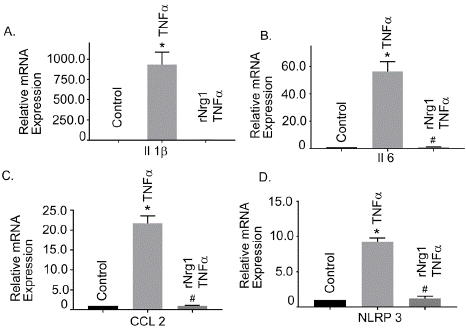
Figure 3: Effect of rNrg1 in Tnfa-induced inflammatory responses in human
brain microvascular endothelial cells. Cells were pretreated with rNrg1 for 2h
and exposed to Tnfa at 25 ng/ml for a period of 24h. The mRNA expression
of (A) Il-1β, (B) Il6, (C) CCL2 and (D) NLRP3 were measured using
reverse transcription-quantitative PCR. These experiments were replicated
three-to-five times, and amplifications were performed and normalized to
GAPDH. Results are expressed as the mean ± SEM from 3 independent
experiments. *P<0.05 vs. untreated cells.
Effects of rNrg1 in Tnfa-stimulated NF-kB Signaling in hBMVECs
To determine further if rNrg1 influenced canonical NF-kB signaling pathway, we examined the gene expression of Rel A (p65 gene) and the translocation of NF-kB-p65 at various doses of Tnfa. The results showed an upregulation of Rel A mRNA in Tnfa treated cells which was significantly reduced in pretreated rNrg1 cells (Figure 4A). Next, we determined nuclear translocation of NF-kB-p65 at various time points as shown in Figure 4B. To determine whether rNrg1 pretreatment inhibited the translocation, cells were pretreated with rNrg1 followed by 10 min Tnfa stimulation. Data showed a significant inhibition in rNrg1 treated group (Figure 4C). The NF-kB-p65 translocation is also examined by immunofluorescence cellular staining. We observed that Tnfa treatment facilitates the nuclear translocation of NF-kB-p65 in 10 min as shown green fluorescence inside the nucleus (Figure 4D). However, TNFa induced NF-kB-p65 translocation was significantly blockedin cells pretreated with rNrg1. Together, our data indicated that rNrg1 pre-treatment inhibited Tnfainduced NF-kB activation in hBMVECs.
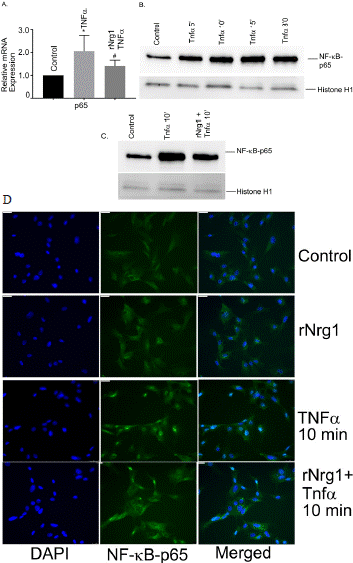
Figure 4: Role of NF-kB in Tnfa-induced inflammatory responses in human
brain microvascular endothelial cells. The mRNA expression of (A) Rel A was
measured using reverse transcription-quantitative PCR. For NF-kB action,
cells were exposed to Tnfa at 25 ng/ml for a period of 5-, 10-, 15 and 30
min and nuclear extracts were prepared. (B) The Western blot analysis was
performed using NF-kB-p65 antibody. The histone (H1) antibody was used
as internal nuclear loading control. (C) Effect of rNrg1 in Tnfa-induced NFkB-
p65 translocation into nucleus. Cells were pretreated with rNrg1 for two
hours and then stimulated with Tnfa at 25 ng/ml for a period of 10 min. The
nuclear extracts were prepared. Western blot analysis was performed using
NF-kB-p65 antibody. These experiments were replicated three-to-five times,
and amplifications were performed and normalized to GAPDH. Results are
expressed as the mean ± SEM from 3 independent experiments. *P<0.05 vs.
untreated cells. (D) Cultured hBMVECs were pre-treated with rNrg1 for 2h
and then stimulated with Tnfa for 10 min. Dual staining immunofluorescence
analysis of NF-kB p65 showed in green. Blue staining was DAPI, for the
nucleus.
Effects of rNrg1 on Tnfa-stimulated Activated Adhesion Molecules in hBMVECs
Adhesion molecules, like ICAM1 and VCAM1 are critical in BBB integrity and are activated in BBB injury. Our study showed that both ICAM1 and VCAM1 were upregulated after Tnfa treatment (Figure 5A and 5B, P<0.05), compared to control cells, respectively. rNrg1 pre-treatment significantly reduced their expression level (Figure 5A and 5B, P<0.05). Our data suggested that rNrg1 may offer protection in Tnfa-induced vascular damaged hBMVECs.
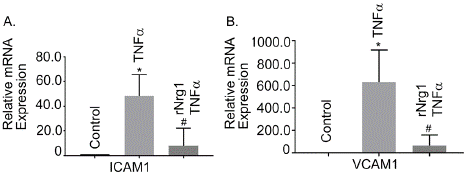
Figure 5: Role of rNrg1 in Tnfa -induced activation of adhesion molecules
in human brain microvascular endothelial cells. Cells were pretreated with
rNrg1 for 2h and exposed to Tnfa at 25 ng/ml for a period of 24h. The mRNA
expression of (A) intercellular adhesion molecule 1(ICAM1) and (B) vascular
cell adhesion protein 1(VCAM1) were measured using reverse transcriptionquantitative
PCR. These experiments were replicated three-to-five times,
and amplifications were performed and normalized to GAPDH. Results are
expressed as the mean ± SEM from 3 independent experiments. *P<0.05 vs.
untreated cells.
Effects of rNrg1 on Tnfa-stimulated Apoptotic Gene Expression in hBMVECs
We evaluated caspase 3 expression in hBMVECs following Tnfa treatment. Tnfa- stimulation significantly increased Caspase 3 mRNA expression compared to untreated cells. rNrg1pre-treatment significantly reduced Caspase 3 gene expression, compared to Tnfatreated cells (Figure 6A, P<0.05). Our data suggested that rNrg1pretreatment protected Tnfa -induced cellular apoptosis in hBMVECs. The protein level of cleaved caspase3 was also determined by Western blotting. The results showed that cleaved caspase 3 was detected after Tnfa stimulation (Figure 6B). rNrg1pre-treatment significantly reduced cleaved Caspase 3 level.
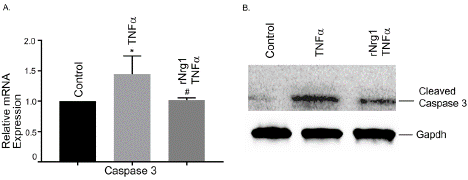
Figure 6: Effect of rNrg1 on Tnfa-stimulated human brain microvascular
endothelial cells. Cells were pretreated with rNrg1 for 2h and exposed to Tnfa
at 25 ng/ml for a period of 12h. The mRNA expression of (A) Caspase 3 was
determined by reverse transcription-quantitative PCR. (B) Representative
image of the Western blot analysis of cleaved Caspase 3 protein level.
These experiments were replicated three-to-five times, and amplifications
were performed and normalized to GAPDH. Results are expressed as the
mean ± SEM from 3 independent experiments. *P<0.05 vs. untreated cells.
Effects of rNrg1 on Neuropilin (Nrp1) and Nerve Growth Factor (NGF) in hBMVECs Stimulated with Tnfa
BBB injury elicits dysregulation of array of several molecules. Here, we examined two important molecules, Nrp1 and NGF in Tnfainduced BBB injury in hBMVECs which are currently unknown. Both candidates are critical in many neurological diseases but their role in neuroinflammation-mediated BBB disruption remain elusive. Our study showed that mRNA expression of NGF was significantly upregulated in Tnfa treated cells and pretreatment of rNrg1 reduced NGF gene expression (Figure 7B). The mRNA expression of Nrp1 was significantly downregulated in Tnfa treated cells and pretreatment of rNrg1 restored the Nrp1 gene (Figure 7A). We further examined Nrp1 at protein level and the data corroborated with mRNA findings as shown in Figure 7C. Tnfa treatment significantly reduced Nrp1 level and pretreatment with rNrg1 restored the Nrp1 level to normal.
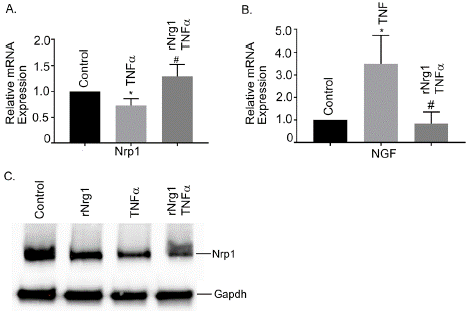
Figure 7: Effect of rNrg1 on Tnfa-stimulated Nrp1 and NGF expression in
human brain microvascular endothelial cells. Cells were pretreated with
rNrg1 for 2h and exposed to Tnfa at 25 ng/ml for a period of 24h. The
mRNA expression of (A) Nrp1 and (B) NGF were measured using reverse
transcription-quantitative PCR. (C) Representative image of the Western blot
analysis of Nrp1 protein. These experiments were replicated three-to-five
times, and amplifications were performed and normalized to GAPD Results
are expressed as the mean ± SEM from 3 independent experiments. *P<0.05
vs. untreated cells.
Effects of rNrg1 in Tnfa-stimulated hBMVECs Monolayer Permeability and TEER Assays
To get an insight about rNrg1’s role in Tnfa-induced increased cellular permeability, hBMVECs were treated with Tnfa for 24h in presence and absence of rNrg1. Tnfa treatment significantly increased the permeability and was significantly reduced with prior treatment of rNrg1 suggesting a protective role in endothelial cell permeability (Figure 8A). Under the similar condition, the TEER assay was determined in hBMVEC monolayers. After treatment with Tnfa for 24h, the TEER in treated cells was significantly decreased as compared to the untreated cells. In a similar set-up, no change in the TEER was observed when treated with rNrg1 prior to Tnfα treatment (Figure 8B). This suggests a protective role of Nrg1 in endothelial cell permeability and barrier integrity during Tnfα induced inflammatory response.
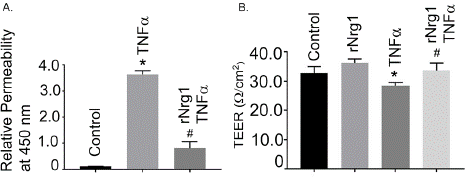
Figure 8: Effect of rNrg1 on Tnfa -stimulated hBMVEC monolayer
permeability assay and TEER analysis. (A) Permeability assays were
performed using Endothelial Trans-well Permeability Assay Kit (cat. No.
CB6929, Cell Biologics, Inc.), according to manufacturer’s protocols. Data
expressed as the A450 absorption readings, which are considered the
relative permeability. Cultured hBMVECs were pre-treated with rNrg1 (100
ng/mL) for 2h and stimulated with Tnfa for 24h. Permeability assays were
performed as described above. (B) Cultured hBMVECs were pre-treated
with rNrg1 (100 ng/mL) for 2h and stimulated with Tnfa for 24h. The TEER
analysis was performed using Millicell® ERS-2 system. The TEER was
measured continuously in hBMVEC with or without rNrg1. The TEER values
are calculated as Ω/cm2. The permeability results are expressed as the mean
± SEM from 3 independent experiments. *P<0.05 vs. untreated cells. The
TEER results are presented as the average ± SEM change of baseline TEER
from at least 3 independent experiments.
Discussion
In the present study, we observed that Nrg1 has the potential to prevent Tnfa-induced BBB damage. The present study also showed that Nrg1 could reduce the inflammatory response by blocking nuclear translocation of NF-kB-65 in hBMVECs. In addition, the present study shows for the first time that NGF and Nrp1 are modulated by Nrg1.
TJ proteins played a crucial role in BBB integrity and maintain a homeostatic balance in the NVU. Deregulation of TJ proteins particularly Claudin 5 and Occludin, are key players in maintaining the integrity of NVU [34,35]. Furthermore, altered permeability across the microvascular environment is a hallmark response in inflammation-mediated pathological conditions [36]. The present study showed that the mRNA expression of Tjp1, Tjp3, Claudin 3, Claudin 5 and Occludin were all significantly downregulated after Tnfa stimulation, which was prevented by Nrg1 pre-treatment. Our observations further support the fact that the presence of proinflammatory cytokines in (Tnfa)-induced hBMVECs lead to the degradation of Occludin and Claudin 5 proteins at the BBB surface and disrupt the TJ structural integrity, while Nrg1 treatment prevented the degradation or damage [37]. Tnfa is a pleiotropic cytokine and its’ response largely exerts via signaling mechanism.
Our study first demonstrated that a significant increase of Rel A (a NF-kB-p65 subunit), NLRP3, Il6, Il1b and CCL2 mRNA expression in Tnfa treated hBMVECs. Interestingly, the enhanced expression of the above genes was significantly reduced in Nrg1 pretreated cells. Our findings of Tnfa induced BBB damage supported previous results observed by other investigators [18-20,38]. However, this is first report that showed a protective role of Nrg1 in inflammation-induced BBB insult. NF-kB is known to be a master regulator of inflammation, which is the key contributor of the pathology of ischemic stroke and BBB injury [39,40]. The nuclear translocation of the NF-kB subunit p65 is a crucial step in NF-kB activation pathway. Tnfa is a potent activator of NF-kB signaling.
The canonical pathway of NF-kB activation is mediated by degradation and phosphorylation of IkBa protein. We determined first the degradation and phosphorylation of IkBa protein in Tnfa treated hBMVECs. Our data showed significant degradation and phosphorylation of IkBa after 5 min treatment of Tnfa, which was restored and reduced by Nrg1 treatment. The observation is confirmed by immunofluorescence microscopy showing the inhibition of NFkB- 65 translocation by Nrg1. The observation suggests that Nrg1 pretreatment provides an anti-inflammatory in hBMVEC remodeling.
Studies have shown that apoptosis might be an outcome of cytokines induced endothelial barrier dysfunction [41,42] whereas others have shown that primary human brain endothelial cells are resistant to apoptosis triggered by Tnfa [43]. In our study, we observed upregulation of the caspase-3 gene and cleaved caspase-3 protein in Tnfa stimulated hBMVEC. Interestingly, Nrg1 treated cells were protected from apoptosis suggesting an anti-apoptotic role of Nrg1. Notably, several reports suggest that caspase-3 is not only an important regulator of cell death by inducing apoptosis but also plays a crucial role in regulating inflammation [44,45]. Our study indicated that Nrg1 may act as an anti-apoptotic molecule as well as inflammation regulator. Future studies are warranted for a detailed mechanism on whether caspase-3 contributes to a role in inflammation-induced cellular death.
It is suggested that inflammation-induced BBB dysfunction facilitates leukocyte migration by activating cellular adhesion molecules in BMVECs causing secretion of proinflammatory molecules, which increase permeability of the vascular wall resulting in BBB dysfunction [46,47]. Our results showed significant upregulation of ICAM1 and VCAM1 gene expression, decreased electrical resistance and increased cellular permeability after Tnfa stimulation and was effectively prevented by pretreatment with Nrg1, indicating a potential role in modulation of adhesion genes.
Finally, our study showed for the first-time involvement of two candidates, the Nrp1 and NGF gene in inflammation-induced BBB disruption. Nrp1 is a single-pass transmembrane, non-tyrosine kinase glycoprotein with a wide variety of physiological functions especially its role in axonal guidance the growth during development as a receptor of semaphoring 3A [48,49]. Although Nrp1 is shown to contribute a role in interferon g-induced inflammatory responses in hBMVECs during BBB injury [50], our study showed that depletion of Nrp1 may be a causal factor for BBB integrity. Additionally, Nrg1 may possibly mitigate the damage by restoring Nrp1 to the basal level. Similarly, NGF has been shown as neuroprotective agent for taming the post-TBI inflammatory condition [51,52], but its’ role in BBB integrity is unknown. Our findings demonstrated that Nrg1 pretreatment reduced the increased NGF expression during inflammatory process suggesting a new role in BBB integrity. These two candidate genes are interesting in BBB integrity, but more studies are warranted to understand their specific role in BBB dysfunction.
To the best of our knowledge, this is the first report describing a possible role of Nrg1 in regulation of TJ components in hBMVECs. Nrg1 is a family member of epidermal growth factor (EGF) and shared EGF-like domain [53]. Nrg1 is reported to contribute protective function in brain ischemia [26-28], however, one report suggested that Nrg1 has beneficial effects on BBB permeability in controlled cortical impact (CCI) rat model, an experimental trauma model, may indicate clinical significance for treatment of CNS injury [49]. Our study corroborated in part of this finding but showed that Nrg1 has the potential in restoring BBB damage.
In conclusion, the present study illustrated that Tnfa-induced BBB dysfunction is associated with significant increase in inflammatory genes, adhesion genes and loss of TJ components including Tjps, Occludin and Claudins which ultimately enhance BBB permeability and dysfunction. Interestingly, the study finds Nrp1 and NGF as possible target genes as their alteration is modulated by Nrg1. BBB damage is effectively restored by Nrg1 treatment suggesting its’ potential as a neuroprotective agent. Furthermore, our study demonstrated that Nrg1 is associated with the regulation of canonical NF-kB signaling pathway, which may open new possibilities for investigating Nrg1 as an anti-inflammatory agent in BBB injury, a critical sequel after TBI. There are limitations in our study. The study utilized a cell line and did not use other cell types surrounding the BBB. We chose hBMVECs as a model system because it is one of the pivotal cells in the NVU which constitute basement membrane and provides structural integrity by forming TJ units and acts as a molecular sieve for the integrity of BBB. Another type of protein known as AJ proteins are also important in TJ organization in BBB maintenance. Our current study did not evaluate AJ protein in BBB dysfunction but is a definitive target for future study. The findings should be corroborated with an in vivo experiment like TBI or CCI. Moreover, our experiments did not delineate the mechanism by which Nrg1 attenuates inflammation, permeability, and restoration of the TJ barrier. Future experiments are warranted to understand the beneficial role of Nrg1 in inflammation and BBB restoration.
Acknowledgement
This material is the result of work with resources and the use of facilities at the VISN 17 Center of Excellence for Research on Returning War Veterans and the Central Texas Veterans Health Care System. The authors acknowledge the Director, Dr. Richard W. Seim, at the VISN 17 Center of Excellence for Research on Returning War Veterans, Waco, Texas, for all the support. The views expressed herein are those of the authors and do not necessarily reflect the official policy or position of the Department of Veterans Affairs or the United States Government.
Funding
The present study was supported by VISN 17 Center of Excellence’s internal funds to Translational Science Core. The authors declared that they do not have any financial or proprietary interest in any material discussed in this article.
Availability of Data and Materials
The data are available from the corresponding author on reasonable request due to privacy/ethical restrictions.
Authors Contributions
SG conceived the experiments. SG and RSG designed the experiments. WS, CH and SG performed the experiments. SG and RSG analyzed the data. SG wrote the manuscript. RSG critically reviewed the manuscript. All authors read and approved the final version of the manuscript.
References
- Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012; 32: 1959–1972.
- Pardridge WM. Molecular biology of the blood-brain-barrier. Mol Biotechnol. 2005; 30: 57–70.
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiology of Disease. 2010; 37: 13–25.
- Tietz S, Engelhardt B. Brain barriers: crosstalk between complex tight junctions and adherens junctions. Journal of Cell Biology. 2015; 209: 493– 506.
- Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun. 2017; 60: 1-12.
- Miller AH, Maletic, V, and Raison, CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry. 2009; 65: 732–741.
- Chodobski A, Zink B, Szmydynger-Chodobska J. Blood-brain barrier changes in traumatic brain injury, in: The Blood-Brain Barrier in Health and Disease. Volume 2, K. Dorovini-Zis (ed). CRC: Boca Raton, FL, pps. 2015; 156–183.
- Yang J, Ran M, Li H, Lin Y, Ma K, Yang Y, et al. New insight into neurological degeneration: Inflammatory cytokines and blood-brain barrier. Front Mol Neurosci. 2022; 5: 1013933.
- Corrigan F, Mander KA, Leonard AV, Vink RJ. Neurogenic inflammation after traumatic brain injury and its potentiation of classical inflammation. Neuroinflammation. 2016; 13: 264.
- Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010; 6: 393- 403.
- Obermeier B, Verma A, Ransohoff, RM. The Blood–Brain Barrier. 2016; 133: 39-59.
- Thal SC, Neuhaus W. The blood-brain barrier as a target in traumatic brain injury treatment Arch. Med. Res. 2014; 45: 698-710.
- Cash A, Theus MH. Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int J Mol Sci. 2020; 21: 3344.
- Pearn ML, Niesman IR, Egawa J, Sawada A, Almenar-Queralt A, Shah SB, et al. Cell Mol Neurobiol. Pathophysiology Associated with Traumatic Brain Injury: Current Treatments and Potential Novel Therapeutics. 2017; 37: 571-585.
- Sulhan S, Lyon KA, Shapiro LA, Huang JH. Neuroinflammation and bloodbrain barrier disruption following traumatic brain injury: Pathophysiology and potential therapeutic targets. J Neurosci Res. 2020; 98: 19-28.
- Kalra S, Malik R, Singh G, Bhatia S, Al-Harrasi A, Mohan S, et al. Pathogenesis and management of traumatic brain injury (TBI): role of neuroinflammation and anti-inflammatory drugs. Inflammopharmacology. 2022; 30: 1153-1166.
- Schimmel SJ, Acosta S, Lozano D. Neuroinflammation in traumatic brain injury: A chronic response to an acute injury. Brain Circ. 2017; 3: 135-142.
- Ni Y, Teng T, Li R, Simonyi A, Sun GY, Lee JC. TNFalpha alters occludin and cerebral endothelial permeability: role of p38MAPK. PLoS One. 2017; 12: e0170346.
- Rochfort KD, Cummins PM. Cytokine-mediated dysregulation of zonula occludens-1 properties in human brain microvascular endothelium. Microvasc. Res. 2015; 100: 48–53.
- Lopez-Ramirez MA, Fischer R, Torres-Badillo CC, Davies HA., Logan K, Pfizenmaier K, et al. Role of caspases in cytokine-induced barrier breakdown in human brain endothelial cells. J. Immunol. 2012; 189: 3130–3139.
- Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003; 284: 14–30.
- Buonanno A. The neuregulin signaling pathway and schizophrenia: from genes to synapses and neural circuits. Brain Res Bull. 2010; 83: 122–131.
- Mei L, Nave KA. Neuregulin ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron. 2014; 83: 27‐49.
- Fazzari P, Paternain AV, Valiente M, Pla R, Lujan R, Lloyd K, et al. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signaling. Nature. 2010; 464: 1376–1380.
- Cui MY, Fu YQ, Li ZL, Zheng Y, Yu Y, Zhang C, et al. Neuregulin-1/PI3K signaling effects on oligodendrocyte proliferation, remyelination and behaviors deficit in a male mouse model of ischemic stroke. Exp Neurol. 2023; 362: 114323.
- Navarro-González C, Huerga-Gómez A, Fazzari P. Nrg1 Intracellular Signaling Is Neuroprotective upon Stroke. Oxid Med Cell Longev. 2019; 9: 3930186.
- Xu Z, Croslan DR, Harris AE, Ford GD, Ford BD. Extended therapeutic window and functional recovery after intraarterial administration of neuregulin-1 after focal ischemic stroke. J Cereb Blood Flow Metab. 2006; 26: 527-535.
- Xu Z, Ford GD, Croslan DR, Jiang J, Gates A, Allen R, Ford BD. Neuroprotection by neuregulin-1 following focal stroke is associated with the attenuation of ischemia-induced pro-inflammatory and stress gene expression. Neurobiol Dis. 2005; 19: 461-470.
- Yang Z, Jiang Q, Chen SX, Hu CL, Shen HF, Huang PZ, et al. Differential changes in Neuregulin-1 signaling in major brain regions in a lipopolysaccharide-induced neuroinflammation mouse model. Mol Med Rep. 2016; 14: 790-796.
- Xu Z, Jiang J, Ford G, Ford BD. Neuregulin-1 is neuroprotective and attenuates inflammatory responses induced by ischemic stroke. Biochem Biophys Res Commun. 2004; 322: 440-446.
- Stewart W, Hejl C, Guleria RS, Gupta S. Effect of thymosin β4 on lipopolysaccharide stimulated brain microvascular endothelial cell remodeling: A possible role in blood brain barrier injury. Exp Ther Med. 2023; 26: 468.
- Kumar S, Gupta S. Thymosin Beta 4 Prevents Oxidative Stress by Targeting Antioxidant and Anti-Apoptotic Genes in Cardiac Fibroblasts. PLoS one. 2011; 6: e26912.
- Li L, Bounds KR, Chatterjee P, Gupta S. MicroRNA-130a, a Potential Antifibrotic Target in Cardiac Fibrosis. J Am Heart Assoc. 2017; 6: e006763.
- Findley MK, Koval M. Regulation and roles for claudin-family tight junction proteins. IUBMB Life. 2009; 61: 431–437.
- Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008; 1778: 660–669.
- Förster C, Burek M, Romero IA, Weksler B, Couraud PO, Drenckhahn D. Differential effects of hydrocortisone and TNF {alpha} on tight junction proteins in an in vitro model of the human blood–brain barrier. J Physiol. 2008; 586: 1937–1949.
- Nishioku T, Matsumoto J, Dohgu S, Sumi N, Miyao K, Takata F, et al. Tumor necrosis factor-alpha mediates the blood–brain barrier dysfunction induced by activated microglia in mouse brain microvascular endothelial cells. J Pharmacol Sci. 2010; 112: 251-254.
- Aslam M, Ahmed N, Srivastava R, Bernhard H. TNF-alpha induced NFkB signaling and p65 (RelA) overexpression repress Cldn5 promoter in mouse brain endothelial cells Cytokine. 2012; 269-275.
- Ridder DA, Schwaninger M. NF-kappaB signaling in cerebral ischemia. Neuroscience. 2009; 158: 995–1006.
- Liu N, Ai-Li L, Xiao-Ping Z, Qiang C, Wei C. P120 catenin attenuates lipopolysaccharide-induced blood-brain barrier dysfunction and inflammatory responses in human brain microvascular endothelial cells. Int. J Clin Exp Pathol. 2015; 8: 4204-4212.
- Basuroy S, Bhattacharya S, Leffler CW, Parfenova H. Nox4 NADPH oxidase mediates oxidative stress and apoptosis caused by TNF-alpha in cerebral vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2009; 296: C422– C432.
- Petrache I, Verin AD, Crow MT, Birukova A, Liu AF, Garcia JGN. Differential effect of MLC kinase in TNF-alpha-induced endothelial cell apoptosis and barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001; 280: L1168–L1178.
- Wosik K, Biernacki K, Khouzam MP, Prat A. Death receptor expression and function at the human blood brain barrier. J. Neurol. Sci. 2007; 259: 53–60.
- Sairanen T, Szepesi R, Karjalainen-Lindsberg ML, Saksi J, Paetau A, Lindsberg PJ. Neuronal caspase-3 and PARP-1 correlate differentially with apoptosis and necrosis in ischemic human stroke. Acta Neuropathol. 2009; 118: 541–552.
- Glushakova OY, Glushakov AA, Wijesinghe DS, Valadka AB, Hayes RL, Glushakov AV. Prospective clinical biomarkers of caspase-mediated apoptosis associated with neuronal and neurovascular damage following stroke and other severe brain injuries: Implications for chronic neurodegeneration. Brain Circ. 2017; 3: 87-108.
- Lee BK, Lee WJ, Jung YS. Chrysin Attenuates VCAM-1 Expression and Monocyte Adhesion in Lipopolysaccharide-Stimulated Brain Endothelial Cells by Preventing NF-kappaB Signaling. Int. J. Mol. Sci. 2017; 18: 1424.
- Zhang Q, Zheng M, Betancourt CE, Liu L, Sitikov A, Sladojevic N, et al. Increase in Blood-Brain Barrier (BBB) Permeability Is Regulated by MMP3 via the ERK Signaling Pathway. Oxid. Med. Cell. Longev. 2021; 6655122.
- He Z, Tessier-Lavigne M. Neuropilin is a receptor for the axonal chemorepellent semaphorin III. Cell. 1997; 90: 739–751.
- Schwarz Q, Vieira JM, Howard B, Eickholt BJ, Ruhrberg C. Neuropilin 1 and 2 control cranial gangliogenesis and axon guidance through neural crest cells. Development. 2008; 135: 1605-1613.
- Wang Y, Cao Y, Mangalam AK, Guo Y, LaFrance-Corey RG, Gamez JD, et al. Neuropilin-1 modulates interferon-gamma-stimulated signaling in brain microvascular endothelial cells. J. Cell. Sci. 2016; 129: 3911–3921.
- Prencipe G, Minnone G, Strippoli R, et al. Nerve growth factor downregulates inflammatory response in human monocytes through TrkA. Journal of Immunology. 2014; 192: 3345–3354.
- Chiaretti A, Conti G, Falsini B, et al. Intranasal nerve growth factor administration improves cerebral functions in a child with severe traumatic brain injury: a case report. Brain Injury. 2017; 31: 1538–1547.
- Lok J, Zhao S, Leung W, Seo JH, Navaratna D, Wang X, Whalen MJ, Lo EH. Neuregulin-1 effects on endothelial and blood-brain-barrier permeability after experimental injury. Transl Stroke Res. 2012; 3 Suppl 1: S119-124.