
Review Article
Austin J Clin Immunol. 2023; 9(2): 1059.
The Current Treatment of Rare Disease, Lysosomal Acid Lipase Deficiency (LAL-D) and Paroxysmal Nocturnal Haemoglobinuria (PNH)
O’Malley S*; Tian F
School of Food Science and Environmental Health, Technological University Dublin (TUD), Ireland
*Corresponding author: O’Malley S School of Food Science and Environmental Health, Technological University Dublin (TUD), Ireland. Tel: 0035312205673 Email: furong.tian@tudublin.ie
Received: June 27, 2023 Accepted: August 01, 2023 Published: August 08, 2023
Abstract
The Paroxysmal Nocturnal Hemoglobinuria (PNH) and Lysosomal acid Lipase Deficiency (LAL-D) are rare disorder disease. Ultomiris and Soliris for PNH and Kanuma for LAL-D are innovative treatments for PNH and LAL-D, respectively. These therapies can treat each ultra-rare condition by alleviating symptoms and lowering mortality rates for both diseases. The aim of this research is to investigate the rare diseases PNH and LAL-D, as well as the existing treatments for both. Each medicine should be safe and have a good therapeutic effect, with a wide range of benefits and few adverse effects. PRISMA (Preferred Reporting Items for Systematic Reviews) was used to evaluate three treatments: Ultomiris and Soliris for PNH and Kanuma for LAL-D. The search was limited to scientific research articles and clinical trials (Publication years between 2007 and 2022). In the analysis, 20 studies were chosen based on their treatment, concentration, population, year of publication, outcomes, and references. Extensive study on the present treatment of PNH and LAL-D was used to draw conclusions on the overall benefits, distribution, efficacy, and safety of the medicines. This study was also utilised to validate the overall improvement in the lives of people with PNH and LAL-D. Ultomiris has an average effect of 96.5% on PNH symptoms, including hemolysis events. Kanuma has a 90% typical impact against LAL-D by reducing symptoms such as LDL-C and normalising Aspartate Aminotransferase (AST) levels (LAL-D symptoms), whilst Soliris has an average 97% effect against PNH, by reducing the presence of lactate dehydrogenase, for example.
Keywords: Rare disease; Lysosomal acid lipase; Deficiency; Paroxysmal; Haemoglobinuria
Introduction
Paroxysmal Nocturnal Hemoglobinuria (PNH) and Lysosomal Acid Lipase Deficiency (LAL-D)
PNH (paroxysmal nocturnal hemoglobinuria) is a rare condition that causes a variety of vague symptoms. It mostly causes problems such as intravascular hemolysis, thrombosis, and bone marrow failure [1]. PNH is a rare blood illness in which blood cells are targeted by a component of the body's immune system. The process of destroying red blood cells is known as "haemolysis," and it is responsible for many of the disease's symptoms.
"Haemolytic PNH" affects approximately 5 persons out of every million in the general population, making it an ultra-rare disease [2]. PNH indications include red/brown/dark urine observed during late-night or early-morning bathroom visits. "Paroxysmal" means "sudden," and "nocturnal" means "at night,". Haemoglobinuria refers to the red blood cells breaking down in blood vessels which eventually appear in the urine [3].
Lysosomal Acid Lipase Deficiency (LAL-D) is an uncommon, chronic, and developing hereditary disease. It inhibits the body's ability to produce the Lysosomal Acid Lipase enzyme (LAL). LAL-D is an extremely rare genetic metabolic condition that causes multiorgan failure and premature infant death in newborns, children, and adults [4]. LAL-D is characterised by cholesteryl esters and triglyceride buildup, primarily in the liver and spleen, although it may occur in other organs [5].
A liver transplant or fibrosis, or both, arise in about 50% of adolescents and adults with LAL-D within three years after the onset of clinical symptoms. The median age of onset for both children and adults with LAL-D is 5.8 years. The LAL-D condition can be identified with a quick blood test [4].
Kanuma (Sebelipase alfa) is an ERT (enzyme replacement therapy). Kanuma's major ingredient, sebelipase alfa, is a synthetic version of the enzyme LAL. Sebelipase alfa substitutes LAL by helping in fat breakdown and avoiding fat accumulation in the body's cells, hence preventing LAL-D symptoms such as liver damage and mortality [6].
Registry and Diagnosis
PNH can be challenging to identify and make a diagnosis because it is an extremely rare condition. Less than 40% of PNH patients have a diagnosis within a year of their first symptoms, and 24% of PNH diagnoses can take five years or more [7].
PNH should be suspected in those who have symptoms of intravascular hemolysis, such as hemoglobinuria or an extremely high serum LDH concentration. A diagnosis may be made using a comprehensive clinical assessment, a complete patient history, and a range of specialty testing [8].
Previously, PNH was diagnosed using tests that sensitised the complement lysis activity on RBCs. In most countries, a Flow Cytometric (FCM) assessment of glycosyl phosphatidylinositol-anchored proteins has essentially replaced the once-common acidified serum lysis test (Ham test). (GPI-AP) [9].
To fully characterise PNH, both bone marrow analysis and flow cytometric detection of GPI expression on peripheral blood cells are required [8].
The Global PNH Patient Registry is a flexible online platform for safely gathering and storing data for medical research. Users of the database can fill out surveys about their own experiences with disease and additionally gain insight about other patients' experiences by viewing compiled survey data [10].
The differential diagnosis of metabolic liver disorders, for instance Lysosomal Acid Lipase Deficiency (LAL-D), can be problematic in clinical settings [11]. Given the disease's unpredictable development and the possibility of liver failure and/or accelerated atherosclerosis being factors in early mortality, there is evidence that LAL-D may be severely underdiagnosed or misdiagnosed [12].
Fortunately, given the availability of a simple diagnostic test and recently approved treatment, LAL-D should be considered in the differential diagnosis in relevant clinical circumstances. An LAL enzyme-based biochemical test can detect LAL-D, allowing for active patient monitoring to identify potential illness outcomes [11].
LAL-D is diagnosed by measurements of activity in the blood or other tissues, as well as assessments of lipid buildup on various organs using radiological techniques such as MRI [13]. The LIPA gene's mutations can also be found using genetic testing. Due to the low level of suspicion for the diagnosis and the commonality of the major clinical and biochemical abnormalities, biopsy findings and radiographic findings are not considered diagnostic, although they do aid increase the suspicion of LAL-D [12].
There are a number of registries for people with LAL-D that gather information, analyse it, and raise awareness of the condition. These registries serve as a catalyst for research that aims to better the lives of PNH patients. These include the Global LAL-D Registry, the LAL-D Alliance Patient Registry, and the LAL-D Registry [14].
Symptoms of PNH and LAL-D
When the immune system targets red blood cells and platelets, PNH occurs. If neglected, PNH can lead to thrombosis, chronic renal failure, or hemolytic anaemia [3]. Hemolysis causes PNH signs and symptoms such as fatigue, dysphagia, dyspnea, stomach pain, erectile dysfunction, haemoglobinuria, and anaemia [15].
Two antibodies, Ultomiris and Soliris, bind to and inactivate the complement protein C5 to prevent blood clotting. Both Soliris and Ultomiris improve PNH patients' quality of life while lowering hemolysis to terminate the symptoms and signs of PNH [16].
Lysosomal Acid Lipase Deficiency (LAL-D) is an uncommon, chronic, and progressive hereditary disease. It inhibits the body's ability to produce the Lysosomal Acid Lipase enzyme (LAL). LAL-D is an extremely rare genetic metabolic condition that causes multiorgan failure and premature infant death in newborns, children, and adults [4]. Sebelipase alfa acts as an LAL substitute by assisting in fat breakdown and limiting fat accumulation in the body's cells, hence preventing LAL-D symptoms such as liver damage and death [6].
Current Treatment
Ultomiris and Soliris as the current treatment for PNH: Eculizumab has been found to improve survival in people with paroxysmal nocturnal hemoglobinuria by decreasing intravascular hemolysis and thrombosis. The goal of raulizumab, a long-acting, second-generation complement component 5 (C5) inhibitor, is to reduce the burden of the eculizumab treatment regimen and the likelihood of breakthrough hemolysis [17]. Ravulizumab binds aggressively and preferentially to the complement protein C5, similarly to the first-generation C5 inhibitor eculizumab, preventing the formation of the terminal complement complex C5b-9, which causes cell lysis [18].
In PNH patients who had previously had eculizumab therapy and were clinically stable, investigations have shown that ravulizumab is noninferior to eculizumab. Nevertheless, Ultomiris may be preferred by patients since it only requires intravenous injections every 8 weeks as opposed to every 2 weeks with Soliris IV injections [19].
The function of ravulizumab and eculizumab in the modulation of complement. The C3 activation stage is the culmination of the complement alternative, lectin, and classical pathways. At this stage, CD55 often suppresses the production of the C3 convertase, preventing the formation of C3b [20]. The membrane assault complex is produced when C5b combines with C3b and other complement proteins after being split into C5a and C5b by the C5 convertase (MAC) [21].
Eculizumab and Ravulizumab both suppress the complement C5 activation [21]. The body's own red blood cells are typically not attacked by the Membrane Attack Complex (MAC), the terminal complement system's by-product, because CD59 inhibits this. Patients suffering PNH however, lack the CD59, allowing MAC to target red blood cells. [22].
The terminal complement system, which includes the synthesis of the MAC, is effectively shut down by both C5 inhibitors' interactions with complement C5 [22]. As a result, PNH patients experience much less hemolysis and a decrease in Neisseria clearance [23].
Kanuma as the Current Treatment for LAL-D
Alexion Pharmaceuticals, Inc developed Sebelipase alfa (Kanuma), a recombinant human Lysosomal Acid Lipase (LAL) for long-term enzyme replacement therapy in patients with LAL deficiency [24]. Kanuma was authorised as an ERT for LAL insufficiency in the European Union and the United States in 2015, respectively [25].
Preclinical research has demonstrated how the creation of an LAL Enzyme Replacement Treatment (ERT) permits the rectification of metabolic flaws cause by LAL-D [26]. In vivo studies in a mouse model, demonstrated how LAL replacement directs the enzyme to the intended compartment by LAL replacement, which decreases the damaging effects of LAL-D. The LAL-deficient invivo model revealed that treated mice outlived untreated animals and that hepatic lipid storage was dramatically enhanced [27].
Ultomiris and Soliris for PNH and Kanuma for LAL-D are innovative treatments for PNH and LAL-D, respectively. These therapies can treat each ultra-rare condition by alleviating symptoms and lowering mortality rates for both diseases. Each medicine should be safe and have a good therapeutic effect, with a wide range of benefits and few adverse effects.
This review is to demonstrate the effects of current PNH and LAL-D treatment by presenting correct data from scientific articles, studies, and clinical trials.
The object will be focus on PNH and LAL-D impact the patient, the efficacy and safety of the current therapeutics for PNH and LAL-D. The global approval of the treatments, and the benefits and drawbacks of the novel medicine will be discussed.
Method
Systematic Reviews and Meta-Analyses
A Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) 2020 checklist was completed, and a flowchart was created in accordance with the PRISMA rules and registration information. The screening process was used to determine articles for inclusion and exclusion. Reasons for articles being excluded varied from; Studies that are not a Randomize Clinical Trial (RCT), Not relevant to the current treatment of PNH and LAL-D, and Language limitation. During the screening process, the eliminated articles, were recorded using the principles outlined in the PRISMA statement 2020 (Figure 2).
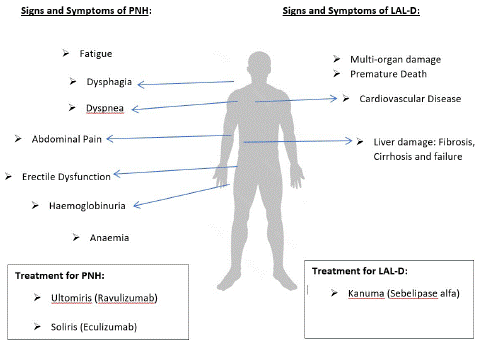
Figure 1: Graphical summary to describe the signs and symptoms of each rare condition, as well as the treatment for the disease.
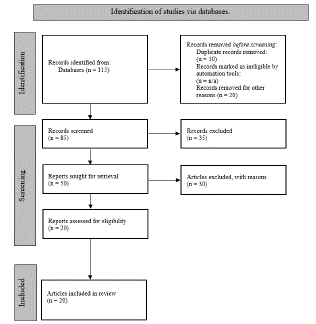
Figure 2: PRISMA flowchart for finding literature.
Global Approval of Ultomiris, Soliris and Kanuma
Ultomiris was authorised by the FDA for adult patients with PNH on December 21, 2019, and for children and adolescents with PNH on June 7, 2021 (Figure 3). Illustrates the nations where Ultomiris, Soliris, and Kanuma have received global approval. Ultomiris was approved in the US, EU, and Japan for the treatment of certain adults with PNH and for certain children with PNH in the US and EU [28]. The first medicine authorised to treat individuals with Paroxysmal Nocturnal Hemoglobinuria (PNH) and lessen hemolysis was Soliris. On Mar 19, 2007, FDA approved Solirs for PNH. In approximately 50 nations throughout the world, including the United States, the European Union, and Japan, Soliris has been given the go-approval for the treatment of PNH patients [29]. European Union, Japan, and the United States have all approved of Kanuma. The first medication authorised for LAL-D patients is Kanuma, which was approved by the FDA on Dec 8, 2015 [4].
Study Findings Following the Use of the Screening Procedure
The information in the table was compiled from key articles about Ultomiris, Soliris, and Kanuma as successful treatments for PNH and LAL-D, with Kanuma successfully reducing LAL-D related symptoms and Ultomiris and Soliris effectively alleviating PNH symptoms. Table 1 contained 20 papers. Treatment, Concentration, Disease, Number of People, Year of Publication, Description, Results, and References were listed in the columns from left to Right.
Each article's information included the number of cohorts that participated in the experiment or article, the amount of the treatment that was used in the clinical trial or article, the year the article or experiment was published, and the results of the treatment based on serious or adverse events. The first informational load focuses on Ultomiris's potential to treat PNH symptoms, including a decrease in plasma Lactate Dehydrogenase (LDH), involved a systematic review with doses ranging from 100 to 900 mg. The second therapy involves the drug soliris, and it involves weight-based dosing or concentrations ranging from 600 to 1,200 mg for the treatment of PNH symptoms such haemoglobin stability at a rate level of 48.8%. The third treatment is related to Kanuma and involved a systematic review with concentrations ranging from 0.35 to 3 mg, easing symptoms of LAL-D such as a reduction in LDL-C and a reduction in non-HDL-C.
Result and Discussion
Research Articles Distribution for the Three Medicines
Twenty publications totalling seven each regarding Ultomiris and treating PNH, six each about Soliris treating PNH, and seven each about Kanuma treating LAL-D were included in the results, as shown in table 1. The percentage of publications featuring Ultomiris, Soliris, and Kanuma is shown in Figure 3.
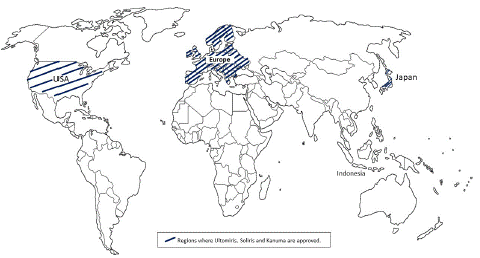
Figure 3: Global approval of Ultomiris, Soliris and Kanuma.
![]()
Concentration
Sample size
Year
Results
Reference
Ultomiris
PNH
1
100 -300 mg
42
2017
Half-life of 32 days
[20]
2
900mg or 1800mg /4 weeks
13
2018
Rapid and sustained LDH (73%-90% at end point vs baseline).
[20]
3
30-mL IV
270
2018
Ravulizumab:
[29]
Affected/at risk at8.80%
Eculizumab;
Affected/ at risk at 7.4%
4
30-mL IV
246
2016
FACIT-F and EORTC-QLQ-C30 scores improved
[39]
5
2700mg-3000mg
191
2021
Common TEAEs for Ultomiris: fatigue (n = 13 [13.5%]),
[40]
6
Weight-based doses
12
2018
Serious adverse events = 2/5
[18]
7
2400 mg / 8 weeks
201
2020
LDH percentage reduction ranging from 72.9% to 89.6%.
[41]
Soliris
PNH
1
600mg-1200 mg
345
2021
11 (3.2%) patients did not improve with Soliris.
[31]
2
N/A
59
2016
History of bone marrow disorder: Yes 4 (7),
[9]
3
600 mg IV /week
176
2017
Death (3)
[42]
4
600 mg IV /week
153
2020
3 thrombotic events in 22 individuals.
[33]
5
900mg / 2 weeks
87
2007
Patients haemoglobin stability at a median rate of 48.8%,
[32]
6
600 mg IV/week
123
2015
Eculizumab-treated patients with fewer thrombotic events (TEs)
[43]
Kauuma
LAL-D
1
Sebelipase Alfa:
66
2015
TEAEs: 6 SAEs: 0
[34]
0.35 -3mg/kg
IRRS: 0 TEAES: 0
2
IV 0.35 -1 mg/kg qow doses
9
2017
TEAEs: 8 SAEs: 1
[44]
IRRS: 2 TEAES: 0
3
IV 1 (mg/kg) qow
19
2022
A median ALT and AT levels decreased by 42.0%.
[35]
4
IV 1 (mg/kg) qow
66
2016
Changes from baseline of ARISE trial:
[45]
a reduction in LDL-C, a reduction in non-HDL-C
5
IV 1 mg/kg /2 weeks,.
21
2022
The first-in-human sebelipase alfa trial, significantly lower ALT and AST values
[26]
6
IV 1 mg/kg /2 weeks,.
110
2022
Tachycardia (70%), pyrexia (60%), irritability (50%), agitation (40%), urticaria (40%)
[25]
7
IV 1 mg/kg /2 weeks,..
3
2018
Stiffness decreased from 8.6 kPa to 7.4 kPa
[46]
Table 1: Experimental research on the Treatment of PNH and LAL-D.
Table 1 lists the average drug efficacy and safety of this systematic review on Ultomiris, Soliris and Kanuma.
The concentrations per ingredient in each research were tabulated and shown in Figure 3. The figures with the title “concentrations” “drug efficacy” “safety” and “benefits and drawbacks” refer to the treatment concentrations and outcomes used in the experiment or article.
![]()
Treatment
Advantages
Reference
Ultomiris (Ravulizumab)
1
Ravulizumab is an antibody that is thought to aid the immune system avoid attacking red blood cells.
[48]
2
Developed into a phase 3 study of weekly self-administered subcutaneous Ultomiris.
[48]
3
Ultomiris is designed to last longer in the body than Soliris, therefore can help patients maintain C5 under control for 8 weeks between infusions,
[29]
4
Ultomiris successfully lowers LDH levels, without needing red blood cell transfusions in patients with PNH
[23]
5
Ultomiris is a monoclonal antibody therapy
[29]
Soliris (Eculizumab)
1
Patient’s experienced significant LDH reduction and clinically noticeable improvement in fatigue compared to untreated patients.
[49]
2
Throughout a course of 26 weeks Soliris treatment resulted in stable haemoglobin levels in 49% of the patients (21 out of 43), eliminating the need for red blood cell transfusions.
[37]
3
Patients treated with Soliris have an 82% chance of surviving.
[50]
4
Safely used in pregnant PNH patients, which was previously prohibited for PNH patients due to the danger of blood clots.
[51]
5
Soliris is a monoclonal antibody and an immunosuppressant
[37]
Kanuma (Sebelipase alfa)
1
Sebelipase alfa substitutes the lipase acid enzyme, assisting in the breakdown of fats and preventing them from accumulating in the body's cells.
[36]
2
Sebelipase alfa is well tolerated and resulted in sustained improvements in liver and lipid parameters.
[35]
3
Improved survival in infants with LAL-D
[4]
4
Reduces the accumulation of fat that contributes to medical issues like decreased growth, liver damage, and heart issues.
[52]
Table 2: Novel Therapies of PNH and LALD and it advantage.
The deepest blue segment indicates for Kanuma, and 35% of the sources were used to demonstrate how Kanuma affected LAL-D symptoms and signs. The lighter blue represents 30% of the sources utilised to indicate how Soliris affects PNH symptoms as you move clockwise around the image. 35% of the sources gathered were related to Ultomiris, which is represented by the lightest blue segment.
Novel Therapies of PNH and LALD and it Advantage and Disadvantage
Table 3 depicts the benefits and drawbacks of each treatment for each relevant condition, PNH or LAL-D. Each of the three therapies demonstrated a great deal of advantages with minimal drawbacks.
![]()
Treatment
Disadvantages
Reference
Ultomiris (Ravulizumab)
1
Increased risk of serious and sometimes fatal diseases such as meningococcal infections and sepsis.
[47]
2
Cost of treatment
[24]
3
Possible allergic reactions to acrylic adhesive
[54]
4
Prevalent side effects include upper respiratory infection, diarrhoea, nausea, and elevated blood pressure.
[54]
Soliris (Eculizumab)
1
Potentially life-threatening side effects
[35]
2
Variations in response profiles
[35]
3
Cost of treatment.
[35]
4
Potential life-long therapy, administered intravenous infusion by a healthcare professional every 14 days.
[56]
Kanuma (Sebelipase alfa)
1
Clinical data for the indicated populations were sparse and ambiguous, limiting the amount of trust that can be placed in the conclusions of the economic analysis.
[4]
2
Due to the limited duration of the current trials, long-term safety and efficacy for sebelipase alfa are unknown and cannot be proved throughout a lifetime.
[4]
3
Weight gain for patients under the age of 21 years
[4]
4
The utility values employed in the analysis were not derived from LAL deficient patients. Changing the utility values in the pediatric/adult presentations, on the other hand, had no discernible effect on the outcomes.
[4]
Table 3: Novel Therapies of PNH and LALD and it disadvantage.
According to the benefits and downsides of Ultomiris, it helps the immune system and avoids attacking red blood cells. Ultomiris effectively reduced hemolysis in patients, and the drug has a longer dose interval than soliris, improving patient comfort and compliance to treatment. Ultomiris is currently in phase 3 clinical trials for a self-administered subcutaneous dose [29]. Consequently, Ultomiris is more accessible to PNH patients [18]. However, Ultomiris has several drawbacks, including an increased risk of serious and occasionally fatal diseases such as meningococcal infections and sepsis [3]. Upper respiratory infection, diarrhoea, nausea, and increased blood pressure are common adverse effects [30].
Eculizumab/Soliris is the most used medication to treat PNH (Soliris) [8,31]. The study demonstrated that Soliris is well tolerated and found no new safety issues [32,33] compared to untreated patients, Soliris patients exhibited considerable LDH reduction and clinically evident improvement in fatigue [9]. However, Soliris has the potential to be a life-long medication that must be administered intravenously by a healthcare expert every 14 days, which might be inconvenient for patients [33].
Kanuma works by replacing the lipase acid enzyme, assisting in fat breakdown, and improving the lives of people suffering with LAL-D [34,35] was discovered that Kanuma gives infants a better chance of survival. Sebelipase alfa is well tolerated and improves liver and lipid indicators over time [4].
[26]. Clinical evidence for kanuma, on the other hand, suggested some limited trust and ambiguous economic analysis outcomes, as well as long-term safety and efficacy [25]. This is due to the studies' brief duration and limited population size.
The Average Efficacy and Safety of Each Therapy Based on Research Articles and Clinical Trials
Figure 5 clarifies appropriate infusion volumes using weight-based dosing because several articles use weight-based dosing. Figure 5 demonstrates the average drug safety and efficacy, as determined by the findings of academic papers and clinical trials. It shows the method of treatment and the average drug efficacy and safety (%) associated with it. Safety (%) is illustrated dark blue and Drug efficacy (%) is illustrated a lighter blue.
This chart was used to determine the safety and efficacy of the current treatments of LAL-D and PNH. Ultomiris, Soliris, and Kanuma have drug efficacy rates of 96.7%, 97%, and 90%, respectively (Figure 4). Ultomiris has a 96.5% average impact on PNH symptoms, including hemolysis episodes. Kanuma has a 90% typical impact against LAL-D by reducing symptoms for instance LDL-C and normalising levels of AST (LAL-D symptoms) [36] whereas Soliris has an average 97% effect against PNH by, for example, reducing the presence of lactate dehydrogenase (symptoms of PNH) [37] Kanuma received a mean safety rating of 96%, which means that there was a 4% possibility of adverse events related to kauma. similarly, to Soliris which has a 95% average safety level with an average 5% chance of adverse or serious events caused by the treatment. Ultomiris however has a lower average safety of 77%. According to statistics from clinical trials and research papers, ravulizumab has a 23% probability of causing major adverse events on average. Ultomiris can induce more severe adverse effects, such as infusion-related symptoms, including back discomfort, fatigue, or drowsiness. Ultomiris increases patients' risk of contracting meningococcal infections, which can be fatal. Upper respiratory tract infection, diarrhoea, nausea, vomiting, headache, elevated blood pressure, and fever are among side effects related to ravulizumab
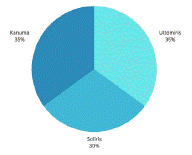
Figure 4: Pie Chart, illustrating the number of research articles used per treatment.
Recommended Infusion Volumes for Each Therapeutic, for the Treatment of PNH and LAL-D
Table 4 displays the infusion volumes indicated for each treatment based on the patient's body weight. Weight-based dosing is used by Ultomiris, Soliris, and Kanuma. The weight-based dosing regimen is helpful in determining the precise infusion volume per patient for a desired therapeutic outcome [38].
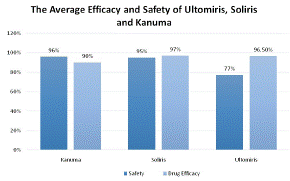
Figure 5: The average outcomes of each research articles based on drug efficacy and safety per treatment.
![]()
Treatment
Disease
Patient Body Weight (kg)
Infusion volumes per body weight
Reference
Ultomiris (Ravulizumab)
PNH
<40
2700mg
[23]
<30
2100mg
<20
600mg
<10
600mg
Soliris (Eculizumab)
PNH
<40
600mg
[37]
<30
600mg
<20
600mg
<10
300mg
Kanuma (Sebelipase alfa)
LAL-D
10-Jan
50 mL per 5mg/kg dose
[36]
24-Nov
150 mL (5mg/kg dose)
25-49
250 mL (5mg/kg dose)
50-99
500 mL (5mg/kg dose)
100-120
600 mL (5mg/kg dose)
Table 4: Recommended infusion volumes for each therapeutic, based on patient body weight.
Table 4 demonstrates how the recommended dose regimen for Ultomiris, Soliris, and Kanuma is determined by the patient's body weight. Ultomiris, for example, the infusion volume for a patient weighing less than 40 kg is 2700 mg, followed by an 8-week maintenance dose. Similarly, to soliris, patients 18 years or older are treated according on their weight; for example, a patient weighing 30 kg is given a 600 mg infusion. Kanuma is calculated by dividing the number of vials to be diluted for infusion by the patient's weight and prescription dose; for example, a patient weighing 1-10 kg should be given 50mL every 5 mg/kg dose.
Limitations
First, this study solely focuses on three medications and overlooks any further treatments that may be available. Second, the study focuses solely on the efficacy and safety of the treatment; it makes no observation of the drug's cost or accessibility in different healthcare settings. Third, because it is based on published literature and clinical trial data, the research may not adequately reflect patient and healthcare provider perspectives or real-world experiences. Furthermore, the study does not consider the potential interactions or adverse effects that may occur when these medications are used in conjunction with other medications. These limitations underscore the need for additional study to investigate alternative treatments, assess real-world outcomes, and consider the broader societal and economic ramifications of these medications.
Conclusions
PRISMA (Preferred Reporting Items for Systematic Reviews) was used to do research on three treatments: Ultomiris, Soliris, and Kanuma. Whereas soliris and ultomiris are used to treat PNH, Kanuma is used to treat LAL-D. The search focused on 20 studies for treatment, concentration, cohort, year of publication, description, findings, and references. The 20 papers concluded that the existing therapies for PNH (Ultomiris and Soliris) and LAL-D (Kanuma) are effective in reducing disease symptoms and signs. Kanuma impacted LAL-D symptoms and signs, according to 35% of the sources. 30% of the sources utilised to demonstrate how Soliris influences PNH symptoms. 35% of the sources consulted referenced Ultomiris. Clinical trials and research publications indicate that the treatment regimen employed to achieve good pharmacological efficacy is based on weight-based dosing. The body weight of the patient determines the recommended dosing regimen for Ultomiris, Soliris, and Kanuma. The infusion volume for Ultomiris, for example, is 2700 mg for a patient weighing less than 40 kg, followed by an 8-week maintenance dose. In conclusion, current PNH (Ultomiris and Soliris) and LAL-D (Kanuma) therapies are undeniably beneficial in providing patients with a virtually normal life expectancy. Ultomiris and Soliris, C5 complement inhibitors, reduce hemolysis, enabling PNH patients to control and suppress symptoms like thrombosis. Drug efficacy, safety, distribution, and advantages all show advances in therapies for both LAL-D and PNH. Ultomiris and Solirs both suppress PNH symptoms and lengthen the lives of PNH patients, with typical drug efficacy rates of 96.5% and 97%, respectively. On the other side, Kanuma assists those with LAL-D. As indicated by its 90% pharmacological efficacy, this ERT effectively and well-tolerated drug to reduce symptoms.
References
- Shah N, Bhatt H. Paroxysmal nocturnal hemoglobinuria. Tampa Florida: Stat Pearls; 2022.
- NHS. What is PNH? Leeds: PNH National Service. Available from: https://www.pnhleeds.co.uk/patients/what-is-pnh/.
- Cleveland Clinic. Paroxysmal nocturnal hemoglobinuria (PNH): symptoms & treatment. Cleveland Clinic; 2022. Available from: https://my.clevelandclinic.org/health/diseases/22871-paroxysmal-nocturnal-hemoglobinuria.
- Alexion Pharmaceuticals. Clinical review report: sebelipase Alfa (Kanuma). Available from: https://www.ncbi.nlm.nih.gov/books/NBK539008/. Alexion Pharmaceuticals, Inc: Indication: Indicated for the treatment of infants, children, and adults diagnosed with lysosomal acid lipase (LAL) deficiency. Ottawa, (ON): Canadian Agency for Drugs and Technologies in Health; 2018.
- Wilson DP, Patni N. Lysosomal acid lipase deficiency. South. MDText.com Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al., editors. Inc. 2000. MA: Dartmouth Publishing. PMID PubMed. Available from: http://www.ncbi.nlm.nih.gov/books/nbk395569/.
- Foundation. Lysosomal acid lipase deficiency. Roche Roche | What is paroxysmal nocturnal haemoglobinuria (PNH)? p. 2023; 2022. Available from: liverfoundation.org. Available from: https://liverfoundation.org/liver-diseases/rare-disease/lysosomal-acid-lipase-deficiency-lald.
- Roche. Roche | What is paroxysmal nocturnal haemoglobinuria (PNH)? www.roche.com. 2022. https://www.roche.com/stories/what-is-pnh/.
- Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005; 106: 3699-709.
- Devos T, Meers S, Boeckx N, Gothot A, Deeren D, Chatelain B, et al. Diagnosis and management of PNH: review and recommendations from a Belgian expert panel. Eur J Haematol. 2018; 101: 737-49.
- Global PNH. Patient registry. Aplastic Anemia and MDS International Foundation; 2023. Available from: https://www.aamds.org/global-pnh-patient-registry.
- Valayannopoulos V, Mengel E, Brassier A, Grabowski G. Lysosomal acid lipase deficiency: expanding differential diagnosis. Mol Genet Metab. 2017; 120: 62-6.
- Strebinger G, Müller E, Feldman A, Aigner E. Lysosomal acid lipase deficiency – early diagnosis is the key. Hepat Med. 2019; 11: 79-88.
- Reiner Ž, Guardamagna O, Nair D, Soran H, Hovingh K, Bertolini S, et al. Lysosomal acid lipase deficiency – an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014; 235: 21-30.
- Committee on diagnostic error in health care, board on health care services, Institute of Medicine, The National Academies of Sciences, Engineering, and Medicine. Improving diagnosis in health care. Washington, (DC): National Academies Press. US; 2015 Balogh EP, Miller BT, Ball JR, editors. Available from: https://www.ncbi.nlm.nih.gov/books/NBK338596/.
- Panse J. Paroxysmal nocturnal hemoglobinuria: where we Stand. Am J Hematol. 2023; 16: S20-32.
- Nishimura JI. Antibody therapy for paroxysmal nocturnal hemoglobinuria. [Rinsho Ketsueki]. Jpn J Clin Hematol. 2020; 61: 929-36.
- Lee SE, Lee JW. Safety of current treatments for paroxysmal nocturnal hemoglobinuria. Expert Opin Drug Saf. 2020; 15: 171-9.
- McKeage K. Ravulizumab: first global approval. Drugs. 2019; 79: 347-52.
- Ann Anderson L. How does Ultomiris compare to Soliris for PNH?; 2022. Drugs.com. Available from: https://www.drugs.com/medical-answers/ultomiris-compare-soliris-pnh-3562113/.
- Gurnari C, Nautiyal I, Pagliuca S. Current opinions on the clinical utility of ravulizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Ther Clin Risk Manag. 2021; 17: 1343-51.
- Connell NT. Ravulizumab: a complementary option for PNH. Blood. 2019; 133: 503-4.
- Dubois EA, Cohen AF. Eculizumab. Br J Clin Pharmacol. 2009; 68: 318-9.
- CZARSKA-THORLEY D. Ultomiris – European Medicines Agency. European Medicines Agency; 2019. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/ultomiris.
- Shirley M. Sebelipase alfa: First global approval. Drugs. 2015; 75: 1935-40.
- Bashir A, Tiwari P, Duseja A. Enzyme replacement therapy in lysosomal acid lipase deficiency (LAL-D): a systematic literature review. Ther Adv Chronic Dis. 2021; 2: 263300402110269:26330040211026928.
- Su K, Donaldson E, Sharma R. Novel treatment options for lysosomal acid lipase deficiency: critical appraisal of sebelipase alfa. Appl Clin Genet. 2016; 9: 157-67.
- Du H, Schiavi S, Levine M, Mishra J, Heur M, Grabowski GA. Enzyme therapy for lysosomal acid lipase deficiency in the mouse. Hum Mol Genet. 2001; 10: 1639-48.
- Vu T, Ortiz S, Katsuno M, Annane D, Mantegazza R, Beasley KN, et al. Ravulizumab pharmacokinetics and pharmacodynamics in patients with generalized myasthenia gravis. J Neurol. 2023; 270: 3129-37.
- Alexion. ALXN1210 (ravulizumab) versus eculizumab in complement inhibitor treatment-naïve adult participants with paroxysmal nocturnal hemoglobinuria (PNH). Full Text View: ClinicalTrials.gov. clinicaltrials.gov; 2023. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT02946463.
- Bektas M, Copley-Merriman C, Khan S, Sarda SP, Shammo JM. Paroxysmal nocturnal hemoglobinuria: role of the complement system, pathogenesis, and pathophysiology. J Manag Care Spec Pharm. 2020; 26: S3-8.
- Cançado RD, Araújo ADS, Sandes AF, Arrais C, Lobo CLC, Figueiredo MS, et al. Consensus statement for diagnosis and treatment of paroxysmal nocturnal haemoglobinuria. Hematol Transfus Cell Ther. 2021; 43: 341-8.
- Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014; 124: 2804-11.
- Bektas M, Copley-Merriman C, Khan S, Sarda SP, Shammo JM. Paroxysmal nocturnal hemoglobinuria: current treatments and unmet needs. J Manag Care Spec Pharm. 2020; 26: S14-20.
- Hoffman EP, Barr ML, Giovanni MA, Murray MF. Lysosomal acid lipase deficiency. Seattle: University of Washington; 2016. Nih.gov. Available from: https://www.ncbi.nlm.nih.gov/books/NBK305870/.
- Burton BK, Sanchez AC, Kostyleva M, Martins AM, Marulkar S, Abel F, et al. Long-term sebelipase Alfa treatment in children and adults with lysosomal acid lipase deficiency. J Pediatr Gastroenterol Nutr. 2022; 74: 757-64.
- Kanuma EMA, European Medicines Agency, European Medicines Agency; 2018. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/kanuma.
- EMA. Soliris – European Medicines Agency. European Medicines Agency; 2018. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/soliris.
- Pan SD, Zhu LL, Chen M, Xia P, Zhou Q. Weight-based dosing in medication use: what should we know? Patient Prefer Adherence. 2016; 10: 549-60.
- Fattizzo B, Cavallaro F, Oliva EN, Barcellini W. Managing fatigue in patients with paroxysmal nocturnal hemoglobinuria: A patient-focused perspective. J Blood Med. 2022; 13: 327-35.
- Kulasekararaj AG, Hill A, Langemeijer S, Wells R, González Fernández FA, Gaya A, et al. One‐year outcomes from a phase 3 randomized trial of ravulizumab in adults with paroxysmal nocturnal hemoglobinuria who received prior eculizumab. Eur J Haematol. 2021; 106: 389-97.
- Griffin M, Kelly R, Pike A. A review of the treatment landscape in paroxysmal nocturnal haemoglobinuria: where are we now and where are we going? Ther Adv Rare Dis. 2020; 1: 263300402095934:2633004020959349.
- SHEPHERD clinical trial. Eculizumab to treat paroxysmal nocturnal hemoglobinuria - https:. Available from: http://classic.clinicaltrials.gov/ct2/show/NCT00130000.
- Loschi M, Porcher R, Barraco F, Terriou L, Mohty M, de Guibert S, et al. Impact of eculizumab treatment on paroxysmal nocturnal hemoglobinuria: a treatment versus no-treatment study. Am J Hematol. 2016; 91: 366-70.
- Clinical trial one (LAL-cl01). Safety, tolerability and pharmacokinetics of SBC-102 (sebelipase Alfa) in adult participants with lysosomal acid lipase deficiency - https:. Available from: http://classic.clinicaltrials.gov/ct2/show/NCT01307098.
- Trial A. Results www.ncbi.nlm.nih.gov. Canadian Agency for Drugs and Technologies in Health; 2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539012/.
- Ashok D, Prasad C, Walsh J, Colaiacovo S, Rupar T. A338 Lysosomal acid lipase deficiency (lal-d): from diagnosis to therapy in Canada. J Can Assoc Gastroenterol. 2018; 1: 488-9.
- Marshall H, Ultomiris BM. Alternatives, cost, side effects, dosage, and more; 2022. Available from: http://www.medicalnewstoday.com. Available from: https://www.medicalnewstoday.com/articles/drugs-ultomiris.
- Alexion Pharmaceuticals. Alexion Announces Phase 3 Study of Weekly subcutaneous ULTOMIRIS® (ravulizumab-cwvz) Met Primary Endpoint | Alexion Pharmaceuticals; 2020. Inc. Available from: media.alexion.com. Available from: https://media.alexion.com/news-releases/news-release-details/alexion-announces-phase-3-study-weekly-subcutaneous-ultomiris.
- Almeida AM, Bedrosian C, Cole A, Muus P, Schrezenmeier H, Szer J, et al. Clinical benefit of eculizumab in patients with no transfusion history in the International Paroxysmal Nocturnal Haemoglobinuria Registry. Intern Med J. 2017; 47: 1026-34.
- Terriou L, Patriquin CJ, Griffin M, Lee JW, Gustovic P, Patel AS, et al. Long-term survival benefit of eculizumab treatment in patients with paroxysmal nocturnal hemoglobinuria: data from the International PNH registry. Blood. 2021; 138: 2188-8.
- Kelly RJ, Höchsmann B, Szer J, Kulasekararaj AG, de Guibert S, Röth A, et al. Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2015; 373: 1032-9.
- Medicines. KANUMA 2mg/ml concentrate solution – Patient Information Leaflet (PIL) – (emc); 2022. Available from: http://www.medicines.org.uk. Available from: https://www.medicines.org.uk/emc/product/7093/pil#gref.
- Alexion Pharmaceuticals, ULTOMIRIS (ravulizumab-cwvz). About ULTOMIRIS.
- MedScape. Soliris (eculizumab) dosing, indications, interactions, adverse effects, and more. 2020;2023:~:text=It%20is%20usually%20given%20every.