
Case Report
Austin J Clin Case Rep. 2025; 12(1): 1351.
Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-Like Episodes (MELAS), in Two Mexican Families
Balderas Correa JJ1, Romo Calvillo HB1, Sánchez Mendoza AE2, Luna Muñoz J3 and Montiel Sosa JF2*
¹Unidad de Investigación Multidisciplinaria Laboratorio 18, FESC, Universidad Nacional Autónoma de México, (UNAM), México
²Departamento de Ciencias Biológicas, FESC, UNAM, México
³Universidad de Pachuca, México
*Corresponding author: Montiel Sosa JF, Unidad de Investigación Multidisciplinaria Laboratorio 18, Universidad Nacional Autónoma de México, México. Carretera Cuautitlán Teoloyucan Km 2.5, San Sebastian Xhala, 54714, Cuautitlán Izcalli, Estado de México, México Tel: +525523995678; Email: fmontiel_sosa@yahoo.com.mx
Received: February 21, 2025; Accepted: March 12, 2025; Published: March 17, 2025;
Abstract
MELAS syndrome is a mitochondrial disorder primarily affecting the nervous and muscular systems, often associated with the m.3243A>G mutation in the MT-TL1 gene. The severity of symptoms depends on heteroplasmy levels, which vary between individuals and tissues. Two Mexican families with suspected MELAS were studied. Peripheral blood samples were analyzed using PCRRFLP. The m.3243A>G mutation was detected in all individuals. In Family 1, the symptomatic son had 17% heteroplasmy, while the asymptomatic mother and brother had 23% and 13%, respectively. In Family 2, the mother and daughter showed 7.7% and 22.8% heteroplasmy, both asymptomatic. Genetic testing is crucial for MELAS diagnosis, even in asymptomatic carriers. The variability in heteroplasmy levels underscores the need for complementary analyses, such as muscle biopsy sequencing, for a more precise diagnosis.
Keywords: MELAS; Mitochondrial DNA; m.3243A>G; Heteroplasmy; Genetic diagnosis
Introduction
Mitochondrial diseases comprise a group of disorders caused by mutations in both mitochondrial DNA (mtDNA) and nuclear DNA (nDNA), affecting the encoding of proteins and essential components for mitochondrial function [1,2].
Mutations in mtDNA can compromise the cell's bioenergetic efficiency, primarily impacting tissues and organs with high energy demands. Among these disorders is mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome, which predominantly affects the nervous and muscular systems [3,4].
The clinical manifestations of MELAS typically appear in early life and are characterized by headaches, muscle weakness, hearing loss, lactic acidosis, and stroke-like episodes. Other reported symptoms include diabetes, vomiting, and short stature [5].
Most MELAS cases are associated with mtDNA mutations, with the m.3243A>G variant being the most frequent. This mutation, located in the MT-TL1 gene, which encodes tRNALeu, is responsible for approximately 80% of cases. Moreover, the expression and severity of the disease depend on the heteroplasmy level, which refers to the proportion of mutant mtDNA relative to the wild type. This percentage varies across tissues and can influence both the onset and severity of symptoms. For this reason, this study presents the clinical cases of two Mexican families with suspected MELAS, in whom the presence of this mutation was identified [3,5].
Case 1
A 50-year-old woman and her two sons, aged 24 and 14, visited the laboratory to evaluate the presence of the m.3243A>G variant associated with MELAS syndrome. As a family history, in 2004, the same study was conducted on the mother's sister, who presented vomiting, seizures, and recurrent headaches. Genetic analysis confirmed the presence of the variant with a 31% heteroplasmy level, along with the identification of the mitochondrial haplogroup B2 [6].
Since the younger son had begun to exhibit characteristic MELAS symptoms, such as muscle weakness and seizures, along with elevated lactic acid levels in laboratory tests, molecular analysis was performed on all three family members. This also allowed for an assessment of possible maternal inheritance of the mutation. For the diagnosis, a peripheral blood sample was obtained, from which DNA was extracted. The mitochondrial genome region containing the m.3243A>G variant was then amplified by PCR. Following PCR, genotyping by restriction fragment length polymorphism (RFLP) analysis was performed. The presence of the mutation in all three family members was confirmed through agarose gel electrophoresis (Figure 1).
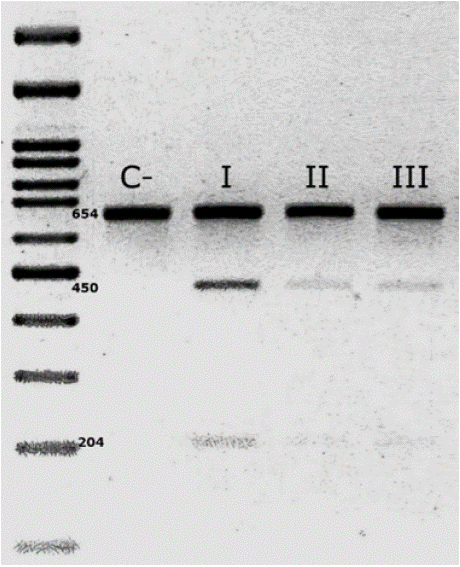
Figure 1: Analysis of the m.3243A>G mutation by 3% agarose gel
electrophoresis using the RFLP technique. Digestion with the ApaI enzyme
generates fragments of 450 and 204 bp in the presence of the mutation,
confirming the variant in the mother (I), the oldest son (II), and the youngest
son (III). In contrast, when the mutation is absent, the PCR-amplified
fragment remains intact, as observed in the negative control (C -).
Additionally, heteroplasmy analysis showed that the mother had 23%, the older son 13%, and the younger son 17%. Despite carrying the variant, the mother and the 24-year-old son have not developed clinical symptoms to date.
Case 2
A 47-year-old woman and her 21-year-old daughter visited the same laboratory to confirm the presence of the m.3243A>G mutation associated with MELAS syndrome. As a relevant family history, the mother's sister had previously been diagnosed with MELAS and passed away after developing the characteristic symptoms of the syndrome, including muscle weakness, diabetes, and recurrent headaches. Despite having reached adulthood without evident clinical signs, both patients expressed concern about the possibility of developing the disease in the future due to their genetic relationship with the affected relative. For molecular diagnosis, DNA was extracted from peripheral blood, and molecular analysis was performed following the same procedure described in Case 1. Genotyping by RFLP and subsequent agarose gel electrophoresis confirmed the presence of the m.3243A>G variant in both patients (Figure 2). Additionally, heteroplasmy quantification revealed 7.7% heteroplasmy in the mother and 22.8% in the daughter.
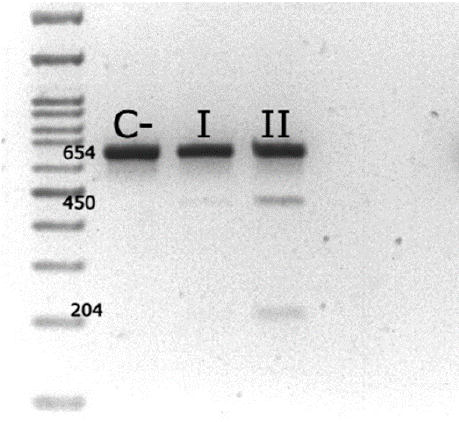
Figure 2: Detection of the m.3243A>G mutation by RFLP analysis in 3%
agarose gel electrophoresis. Digestion with the ApaI enzyme fragments
the PCR product into 450 and 204 bp bands when the mutation is present,
confirming its presence in the mother (I) and her daughter (II). In the absence
of mutation, the amplified fragment remains intact, as observed in the
negative control (C -).
Discussion
Mitochondrial diseases exhibit a wide range of clinical phenotypes resulting from mitochondrial dysfunction in cells of organs with high energy demands. Given the complexity of these disorders and their association with mtDNA mutations, an accurate diagnosis is essential, with genetic testing serving as a key confirmatory tool [7,8].
In the analyzed cases, all members of both families tested positive for the m.3243A>G variant, indicating a potential risk of developing MELAS syndrome. Although most individuals evaluated did not present characteristic symptoms, it is important to consider that disease expression may manifest in adulthood. Furthermore, due to the maternal inheritance of mtDNA, there is a hereditary risk that could affect future generations [3].
Although the heteroplasmy levels detected in the analyzed samples were relatively low, it is crucial to highlight that this proportion varies among individuals, tissues, organs, and cells. Even if the observed values do not exceed the biochemical threshold required to trigger disease symptoms, the possibility of future clinical manifestations cannot be ruled out [9].
Notably, in Case 1, where the younger son has already developed MELAS-compatible symptoms, genetic testing from a muscle biopsy is recommended, as this tissue provides clearer evidence of heteroplasmy levels.
Additionally, in cases where mitochondrial disease remains clinically suspected, whole mitochondrial genome sequencing could be performed to rule out other potential mutations associated with the patient’s condition [7,9].
References
- Habbane M, Montoya J, Rhouda T, Sbaoui Y, Radallah D, Emperador S. Human Mitochondrial DNA: Particularities and Diseases. Biomedicines. 2021; 9: 1364.
- Russell OM, Gorman GS, Lightowlers RN, Turnbull DM. Mitochondrial Diseases: Hope for the Future. Cell. 2020; 181: 168–188.
- Tetsuka S, Ogawa T, Hashimoto R, Kato H. Clinical features, pathogenesis, and management of stroke-like episodes due to MELAS. Metab Brain Dis. 2021; 36: 2181–2193.
- van der Bliek AM, Sedensky MM, Morgan PG. Cell Biology of the Mitochondrion. Genetics. 2017; 207: 843–871.
- El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Molecular Genetics and Metabolism. Academic Press Inc. 2015; 116: 1–2.
- Delgado Sánchez R, Zárate Moysen A, Monsalvo Reyes A, Herrero MD, Ruiz Pesini E, López Pérez MJ, et al. Encefalomiopatía mitocondrial, acidosis láctica y accidentes cerebrovasculares (MELAS) con la mutación A3243G en el gen ARNt Leu(UUR) del ADNmt en el haplogrupo B2 nativo americano. Rev Neurol. 2007; 44: 18-22.
- Davis RL, Liang C, Sue CM. Mitochondrial diseases. In: Handbook of Clinical Neurology. Elsevier B.V. 2018; 147: 125–141.
- Rossmann MP, Dubois SM, Agarwal S, Zon LI. Mitochondrial function in development and disease. Dis Model Mech. 2021; 14: dmm048912.
- Baek MS, Kim SH, Lee YM. The usefulness of muscle biopsy in initial diagnostic evaluation of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. Yonsei Med J. 2019; 60: 98–105.