
Research Article
Austin J Cerebrovasc Dis & Stroke. 2021; 8(1): 1087.
Bioinformatic Identification of Differentially Expressed Genes and Pathways in Intracranial Aneurysm
Tian Q, Han S, Zhang W, Gong P, Xu Z, Chen Q and Li M*
Department of Neurosurgery, Renmin Hospital of Wuhan University, China
*Corresponding author: Mingchang Li, Department of Neurosurgery, Renmin Hospital of Wuhan University, 99-Ziyang Road, Wuhan, Huibei Province, 430060, China
Received: February 27, 2021; Accepted: March 24, 2021; Published: March 31, 2021
Abstract
Background: Intracranial Aneurysm (IA) is a serious disease with high mortality and high morbidity rates, but the pathophysiological mechanisms of IA remain unclear. This study aimed to identify the Differentially Expressed Genes (DEGs) between IA tissues and Superficial Temporal Artery (STA) tissues using bioinformatic analysis.
Methods: To investigate the key genes that are important for IAs, we analyzed microarray datasets (GSE75436) from the Gene Expression Omnibus (GEO) database, including 15 IA samples and 15 normal STA samples. First, we used the GEO2R tool to screen for DEGs (P-value<0.01 and |log2 FC| ≥2) between IA and STA tissues. Subsequently, the Database for Annotation, Visualization, and Integrated Discover software was used to perform function and pathway enrichment analyses. Finally, protein-protein interaction network analysis was performed using the Search Tool for Retrieval of Interacting Genes and Cytoscape software. Real-Time Quantitative Polymerase Chain Reaction (RT-QPCR) was performed to prove our assumption.
Results: A total of 829 DEGs, of which 399 were upregulated and 430 were downregulated, were identified. The upregulated genes were mostly associated with Staphylococcus aureus infection, amoebiasis, rheumatoid arthritis, phagocytosis, and tuberculosis. The downregulated genes were mainly involved in vascular smooth muscle contraction, calcium signaling, histidine metabolism, cGMP-PKG signaling, and cAMP signaling. From the DEGs, five genes were selected as hub genes on the basis of the connection degree, which is one of 12 calculation methods from a plugin of Cytoscape called cytoHubba. The PCR results demonstrated that the expression levels of the top five hub genes, namely, Tumor Necrosis Factor (TNF), interleukin 8 (IL-8), Protein Tyrosine Phosphatase Receptor Type C (PTPRC), interleukin 1β (IL-1β), and Toll-like receptor 4 (TLR 4), were significantly higher in the IA samples than in the STA samples.
Conclusion: TNF showed higher expression in the IA samples than in the STA samples. Thus, this gene may be involved in the occurrence and development of IA. The immune response and inflammation play important roles in the progression of IA. However, the specific pathophysiological mechanism needs further study.
Keywords: Bioinformatics; differentially expressed genes; pathway; intracranial aneurysm
Introduction
Intracranial Aneurysms (IAs) are abnormal protrusions occurring on the wall of the intracranial arteries, with a prevalence of 3% to 5% in the general population; the risk of rupture is nearly 1% [1,2]. The rupture of IAs causes aneurysmal Subarachnoid Hemorrhage (aSAH), which has high mortality and high morbidity rates. Despite immediate treatment upon diagnosis, aSAH is still fatal in 30-40 % of cases [3,4]. Treatment of IAs before rupture has been advocated for reducing its morbidity and mortality. With the development of neuroimaging techniques, patients with incidental IAs are identified more frequently. While the mechanism of aneurysm formation is still poorly understood by researchers, many factors, such as older age, hypertension, smoking, larger size of aneurysm, family history of IAs and irregular morphology, have been demonstrated to be involved in the increased formation of aneurysms [5]. However, there are still some limitations to those predictive models due to the incomplete understanding of the true molecular mechanisms responsible for aneurysm formation. More insight into the pathomechanism of aneurysm formation and development will facilitate clinical management choices and provide more treatment options in the future. Therefore, the pathogenesis of IAs must be elucidated, and molecular markers for the diagnosis and screening of IAs must be identified to provide a new target for the prevention and treatment of IAs.
Several studies have indicated that many genes are involved in the progression of IAs. For example, in humans, genes related to the muscle system and cell adhesion in IA are downregulated compared with those in normal intracranial arteries. The immune/ inflammatory system has been reported to be associated with IAs, but the exact mechanism remains to be elucidated [3]. Another study provided strong evidence for MHC class II gene overexpression in human IA tissue and demonstrated that antigen-presenting cells (macrophages, monocytes) play a key role in IA formation [6]. The regulation of apoptosis-related genes, such as NOL1; immune system development-associated genes, such as CD40LG and CD40; and neuron projection development-associated genes, such as STRN, in vascular smooth muscle cells may be involved in the formation of IAs [7]. However, the current study only partially reported the mechanism of IA, and more research on IA is required.
In the present study, we analyzed a microarray dataset (GSE75436) containing 15 IA tissues and 15 STA tissues using a series of bioinformatic methods to discover key IA-related genes, which will provide a potential therapeutic strategy and offer a direction for further research.
Materials and Methods
Source of gene expression data
We downloaded the GSE75436 gene expression profile of IA from the National Center of Biotechnology Information (NCBI) Gene Expression Omnibus (GEO, www.ncbi.nlm.gov/geo). GSE75436 was based on the GPL570 platform ([HG-U-133_PLUS_2] Affymetrix Human Genome U133 Plus 2. 0 Array), and 30 samples were obtained from the database, including 15 IA samples and 15 STA samples. All samples were utilized in our study.
Data processing of Differentially Expressed Genes (DEGs). GEO2R (www.ncbi.nlm.gov/geo2r/) is an online analysis tool that allows users to compare multiple groups of samples in a GEO series. It was used to analyze most GEO series data with gene symbols to identify the DEGs between IA and STA samples. Adjusted P-value<0.01 and |log2fold change| >2 were set as screening thresholds in this study.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of DEGs
The Database for Annotation, Visualization, and Integrated Discovery (DAVID), which is a common tool for online biological analysis, was used to analyze GO annotation and KEGG pathway enrichment of the DEGs in the study. The GO and KEGG pathway analyses of DEGs utilized a cut-off criterion of P<0.01. DAVID was used to analyze the core Biological Processes (BPs), Molecular Functions (MFs), Cellular Components (CCs), and pathways among these DEGs.
Network construction of a Protein-Protein Interaction (PPI) network
PPI network analysis is helpful for studying the molecular mechanism of disease and discovering new drug targets from a systematic perspective. The interaction among proteins encoded by DEGs in this study was analyzed with the Search Tool for the Retrieval of Interacting Genes (STRING; www.string-db.org) tool. An interaction score >0.4 was used to select upregulated and downregulated genes. Subsequently, Cytoscape software was used to visualize the data downloaded from the STRING database. For further analysis, Cytoscape software was utilized to calculate the protein nodes. In this study, 10 genes were selected from the DEGs as hub genes on the basis of the connection degree, which is one of 12 calculation methods from a plugin of Cytoscape called cytoHubba.
Sample preparation and RNA isolation
This study was approved by the Ethics Committee of Renmin Hospital of Wuhan University (Ethical Application Ref: WDRY2019-K090). IA tissues were acquired from patients who underwent microneurosurgery at Renmin Hospital of Wuhan University from 2018 to 2019. Matched Superficial Temporal Artery (STA) tissues were obtained from each patient as a control. Informed consent was signed by all patients. The Institutional Ethics Committee of Renmin Hospital of Wuhan University approved this study in accordance with the principles presented in the Declaration of Helsinki. The tissue samples comprised three IA tissues and three STA tissues. Collected tissues were immediately washed with saline and then transferred to a liquid nitrogen tank and stored at -80ºC until RNA extraction. RNA was isolated from IA and STA tissues by using a TRIzol-based RNA isolation protocol.
Quantification of RNAs by quantitative polymerase chain reaction
RNA was quantified using a NanoDrop spectrophotometer, and 10 ng of the total RNA was reverse transcribed using the TaqMan RNA Reverse Transcription Kit (Applied Biosystems) in accordance with the manufacturer’s protocol. The top five genes (TNF, IL-8, PTPRC, IL-1β, and TLR4) were detected using TaqMan RNA assays (Applied Biosystems) on a 7500 HT quantification real-time polymerase chain reaction instrument (Applied Biosystems). GAPDH was used as an internal control. For all RNAs, a Ct value >35 was defined as undetectable. Relative mRNA expression levels were analyzed by the 2-ΔΔcycle threshold method.
Results
Data of DEGs
On the basis of the previous standard (adjusted P-value <0.01 and |log2fold change| >2), 829 DEGs, of which 399 were upregulated and 430 were downregulated, were identified between the IA and STA samples in our study. The volcano plots (Figure 1A) showed the DEGs were able to distinguish between the IA and STA samples. The top 10 upregulated and downregulated genes are shown in Table 1.
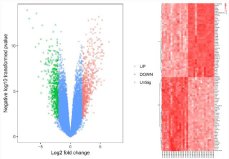
Figure 1: (A) Volcano plot, red dot represent high expression, green represent low expression; (B) hierarchical clusteranalysis of the DEGs, crimson genes with
high expression and light red show cell with low expression. DEGs, differentially expressed genes.
![]()
ID
Gene. symbol
Gene. title
P. Value
logFC
37892_at
COL11A1
collagen type XI alpha 1 chain
1.18E-14
7.24
212942_s_at
CEMIP
cell migration inducing hyaluronan binding protein
1.12E-12
6.27
223642_at
ZIC2
Zic family member 2
2.73E-14
5.66
229290_at
DAPL1
death associated protein like 1
2.54E-07
5.34
1554195_a_at
C5orf46
chromosome 5 open reading frame 46
5.43E-09
4.96
209921_at
SLC7A11
solute carrier family 7 member 11
1.84E-11
4.96
208606_s_at
WNT4
Wnt family member 4
1.57E-05
4.91
204416_x_at
APOC1
apolipoprotein C1
6.82E-08
4.81
1558930_at
LINC00460
long intergenic non-protein coding RNA 460
5.55E-13
4.7
236028_at
IBSP
integrin binding sialoprotein
2.49E-06
4.61
242680_at
AVPR1A
arginine vasopressin receptor 1A
6.60E-11
-5.26
205440_s_at
NPY1R
neuropeptide Y receptor Y1
3.15E-09
-5.26
207317_s_at
CASQ2
calsequestrin 2
1.32E-09
-5.27
206898_at
CDH19
cadherin 19
2.60E-10
-5.28
210135_s_at
SHOX2
short stature homeobox 2
1.44E-13
-5.3
239921_at
COL28A1
collagen type XXVIII alpha 1 chain
6.22E-11
-5.35
220276_at
RERGL
RERG like
5.61E-10
-5.36
223597_at
ITLN1
intelectin 1
1.26E-09
-5.54
205278_at
GAD1
glutamate decarboxylase 1
6.53E-07
-5.64
232054_at
LOC101926951///PCDH20
uncharacterized LOC101926951///protocadherin 20
3.58E-11
-6.46
Table 1: The top ten up-regulated and down-regulated DEPs.
Clustering of DEGs
The 829 DEGs were subjected to clustering analysis using Morpheus software for versatile matrix visualization and analysis. The DEGs between the IA and STA samples were divided into distinct groups through hierarchical clustering on the basis of their expression patterns (Figure 1B).
GO function enrichment analysis of DEGs
GO results are classified into three parts (BP, MF, and CC). Therefore, we analyzed the DEGs in terms of BP, CC, and MF. With the standard of P<0.01, the upregulated DEGs were significantly enriched in BPs including inflammatory response, immune response, cell adhesion, extracellular matrix organization, and neutrophil chemotaxis, whereas plasma membrane, extracellular region, extracellular space, integral component of plasma membrane, and collagen trimer were in the enriched CCs. Enriched MFs were mostly related to chemokine activity, extracellular matrix structural constituent, carbohydrate binding, receptor activity, and IgG binding (Figure 2A; Table 2). In addition, the downregulated DEGs were significantly enriched in BPs including those related to muscle contraction, nervous system development, cell adhesion, neurotransmitter catabolic process, and central nervous system development. Furthermore, DEGs were enriched in CCs including the Z disc, proteinaceous extracellular matrix, actin cytoskeleton, sarcolemma, and sarcoplasmic reticulum membrane. Enriched MFs included actin binding, structural constituent of muscle, cytoskeletal protein binding, primary amine oxidase activity, and ion channel binding (Figure 2B; Table 2).
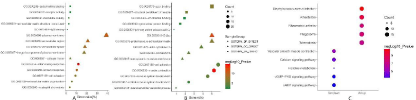
Figure 2: Gene ontology annotation in MF, CC, and BP of (A) up-regulated DEGs and (B) down-regulated DEGs. (C)KEGG enrichment analysis. The functionally
enriched pathways of up-regulated and down-regulated DEGs. KEGG, Kyoto Encyclopedia of Genes and Genomes. DEGs, differentially expressed genes. MF,
molecular function. CC, cellular component. BP, biological process.
![]()
Up-regulated
Sample Group
Q value
Count
Description
GOTERM_BP_DIRECT
8.26E-20
47
GO: 0006954~inflammatory response
GOTERM_BP_DIRECT
3.89E-13
41
GO: 0006955~immune response
GOTERM_BP_DIRECT
2.36E-08
36
GO: 0007155~cell adhesion
GOTERM_BP_DIRECT
8.11E-07
22
GO: 0030198~extracellular matrix organization
GOTERM_BP_DIRECT
9.59E-07
14
GO: 0030593~neutrophil chemotaxis
GOTERM_CC_DIRECT
1.49E-14
154
GO: 0005886~plasma membrane
GOTERM_CC_DIRECT
8.77E-13
83
GO: 0005576~extracellular region
GOTERM_CC_DIRECT
2.11E-11
72
GO: 0005615~extracellular space
GOTERM_CC_DIRECT
6.46E-09
69
GO: 0005887~integral component of plasma membrane
GOTERM_CC_DIRECT
4.42E-07
16
GO: 0005581~collagen trimer
GOTERM_MF_DIRECT
5.68E-04
10
GO: 0008009~chemokine activity
GOTERM_MF_DIRECT
0.008670252
10
GO: 0005201~extracellular matrix structural constituent
GOTERM_MF_DIRECT
0.011087553
16
GO: 0030246~carbohydrate binding
GOTERM_MF_DIRECT
0.037224871
16
GO: 0004872~receptor activity
GOTERM_MF_DIRECT
0.061602241
5
GO: 0019864~IgG binding
Down-regulated
Sample Group
Q value
Count
Description
GOTERM_BP_DIRECT
3.16E-15
22
GO: 0006936~muscle contraction
GOTERM_BP_DIRECT
2.47E-05
19
GO: 0007399~nervous system development
GOTERM_BP_DIRECT
6.84E-05
24
GO: 0007155~cell adhesion
GOTERM_BP_DIRECT
4.31E-04
4
GO: 0042135~neurotransmitter catabolic process
GOTERM_BP_DIRECT
7.66E-04
10
GO: 0007417~central nervous system development
GOTERM_CC_DIRECT
1.88E-13
21
GO: 0030018~Z disc
GOTERM_CC_DIRECT
4.46E-07
21
GO: 0005578~proteinaceous extracellular matrix
GOTERM_CC_DIRECT
3.22E-05
16
GO: 0015629~actin cytoskeleton
GOTERM_CC_DIRECT
4.75E-05
10
GO: 0042383~sarcolemma
GOTERM_CC_DIRECT
4.95E-05
7
GO: 0033017~sarcoplasmic reticulum membrane
GOTERM_MF_DIRECT
7.29E-07
21
GO: 0003779~actin binding
GOTERM_MF_DIRECT
1.42E-06
9
GO: 0008307~structural constituent of muscle
GOTERM_MF_DIRECT
0.002509106
6
GO: 0008092~cytoskeletal protein binding
GOTERM_MF_DIRECT
0.005554401
3
GO: 0008131~primary amine oxidase activity
GOTERM_MF_DIRECT
0.007204843
8
GO: 0044325~ion channel binding
Table 2: Go enrichment analysis of upregulated and downregulated DEGs.
KEGG pathway enrichment analysis of DEGs
In total, 83 pathways, of which 49 were upregulated and 34 were downregulated, were significantly enriched in the DEGs (P<0.01). The upregulated pathways included those involved in Staphylococcus aureus infection, amoebiasis, rheumatoid arthritis, phagocytosis, and tuberculosis. The downregulated genes were mainly correlated with vascular smooth muscle contraction, calcium signaling, histidine metabolism, cGMP-PKG signaling, and cAMP signaling (Figure 2C; Table 3).
![]()
Sample Group
Gene Ratio
P value
Count
Description
IA-up
3.78788
1.35E-11
15
hsa05150: Staphylococcus aureus infection
IA-up
4.54545
3.00E-10
18
hsa05146: Amoebiasis
IA-up
3.78788
1.34E-08
15
hsa05323: Rheumatoid arthritis
IA-up
4.79798
1.50E-08
19
hsa04145: Phagosome
IA-up
4.79798
1.46E-07
19
hsa05152: Tuberculosis
IA-down
2.86396
6.96E-06
12
hsa04270: Vascular smooth muscle contraction
IA-down
3.10263
6.85E-05
13
hsa04020: Calcium signaling pathway
IA-down
1.19332
6.32E-04
5
hsa00340: Histidine metabolism
IA-down
2.6253
6.64E-04
11
hsa04022: cGMP-PKG signaling pathway
IA-down
2.86396
7.06E-04
12
hsa04024: cAMP signaling pathway
Table 3: The top 5-upregulated and downregulated pathway enriched in KEGG.
PPI network construction and hub gene identification
STRING was used to predict PPIs among the DEGs. We constructed the PPI network of the top 10 genes with neighbors ranked by degree of connection, and the resulting network included 192 nodes and 1365 edges (Figure 3A). We also identified the top 5 genes with the shortest path (Figure 3B), which showed the top 5 hub genes that were selected, including TNF, IL-8, PTPRC, IL-1β, and TLR4.
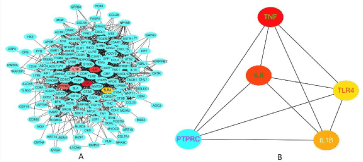
Figure 3: Protein-protein interaction network. This figure demonstrates the large scale and connectivity of the protein-protein interaction network; the nodes in the
figure represent proteins, and the edges are interaction relations. (A) DEGs interact with each other, (B) The most connected 5 genes interact each other calculated
by cytoscape software.
QRT-PCR for validation
To ensure the credibility of the GSE75436 microarray and further validate our results, we reidentified the top 5 hub genes via quantitative Real-Time Polymerase Chain (qRT-PCR) in vitro. The qRT-PCR results demonstrated that the gene expression levels of TNF, IL-8, PTPRC, IL-1β, and TLR4 were significantly higher in the IA group than in the STA group (Figure 4).
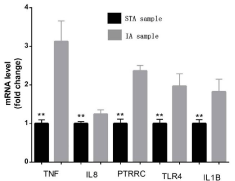
Figure 4: Validation of the top 5 hub genes between IAs tissues and STA
tissues. TNF, IL-8, PTPRC, IL-1β and TLR4 were significantly up-regulated in
IAs tissues. **represent P<0.05 versus STA tissues. Gray graphics represent
STA group, black graphics represent IAs group.
Discussion
IA can cause fatal SAH once ruptured. Symptomatic vasospasm usually occurs 3 days after aSAH, reaching its peak on the 7th day and continuing for 2-3 weeks [8,9]. The overall incidence of IA is 4-6 %, and asymptomatic IA is at least 10 times more prevalent than ruptured IA [10]. IA has high mortality and morbidity rates, and the risk of poor outcome increases because of initial hemorrhage, recurrent hemorrhage, or symptomatic vasospasm [11]. Nevertheless, IAs cannot be examined by routine physical examinations, and cranial computed tomography and digital subtraction angiography are inadequate for the identification of an asymptomatic aneurysm. Therefore, the detection of crucial genes that are involved in IAs would be critical for early diagnosis. In the present study, we recognized 829 DEGs, of which 399 were upregulated and 430 were downregulated, between IA and STA samples. We used GO function and KEGG pathway analyses for further analyses of the DEGs. The upregulated DEGs were related to S. aureus infection, amoebiasis, rheumatoid arthritis, phagocytosis, and tuberculosis, while the downregulated genes were mainly involved in vascular smooth muscle contraction, calcium signaling, histidine metabolism, cGMP-PKG signaling, and cAMP signaling. We selected the top 10 DEGs as hub genes.
Hub genes as candidates for the diagnosis of IAs
In our study, we selected 829 DEGs between IA and STA samples and 10 hub genes in IAs, which were the core nodes in the PPI network. Some of the genes have been reported to be closely related to IAs. Toll-Like Receptors (TLRs) are responsible for the detection of characteristic molecules of pathogens and stimulate the innate immune response. Recent studies showed that TLR4 is associated with the pathogenesis of many inflammatory diseases [12]. The upregulation of TLR2 suggests that this gene is involved in the pathogenesis of IAs [13]. TLR2 and TLR4 can stimulate the proliferation of vascular smooth muscle cells, suggesting that TLR2 and TLR4 play crucial roles in the pathophysiology of IAs [13,14]. A study in a Polish population reported that IL-1β rs16944 is involved in IAs and that IL-1β is upregulated in IAs compared with controls [15]. In addition, the concentration of IL-8 secreted by endothelial cells and leukocytes (including macrophages) in the wall of aneurysms is higher in the lumen of human IAs than in femoral arteries [16]. Another study also found that CSF IL-8 concentration was significantly correlated with the size of IAs, which implied that IL-8 participates in the formation and development of IAs [17]. There is no study about the relationship between the PTPRC gene and IAs.
TNF is essential for IAs
TNF encodes a multifunctional proinflammatory cytokine that belongs to the TNF superfamily. This cytokine is mainly secreted by macrophages. It can bind to its receptors TNFRSF1A/TNFR1 and TNFRSF1B/TNFBR and thus function through them. This cytokine is involved in the regulation of a wide spectrum of BPs, including cell proliferation, differentiation, apoptosis, lipid metabolism, and coagulation. This cytokine has been implicated in a variety of diseases, including autoimmune diseases, insulin resistance, and cancer. Knockout studies in mice also suggested the neuroprotective function of this cytokine. TNF is one of many proinflammatory cytokines that are involved in the inflammatory cascade and can respond to various forms of stress (infection-related, chemical, and mechanical) [18,19]. TNF-α have suggested that inflammation is associated with cerebral aneurysms formed by numerous mediators [20,21]. In addition, TNF-α is overexpressed in patients with SAH compared to healthy controls [22]. It also induces the phenotypic modulation of brain Smooth Muscle Cells (SMCs) through myocardin- and KLF4- regulated pathways. These results demonstrate that TNF-α plays a novel role in promoting a pro-inflammatory or matrix-remodeling phenotype that has important implications for the mechanisms behind IA formation [23]. TNF-α is also upregulated in other aneurysms. For example, inhibition of TNF-α expression by infliximab reduces aneurysm formation and inhibits aneurysm growth in a mouse model of abdominal aortic aneurysm while repressing metalloproteinase expression and macrophage infiltration in aortic tissue [24]. Patients with more than one aneurysm display higher levels of TNFα than those with just one aneurysm [25]. These data suggest that TNF-α plays a crucial role in cerebral aneurysm.
Immune response and inflammation play an important role in the progression of IAs
The top five genes were enriched in S. aureus infection, amoebiasis, rheumatoid arthritis, phagocytosis, and tuberculosis, all of which are related to the immune response and inflammation. Many assumptions have been proposed to elucidate the progression of IAs; one hypothesis is that the inflammatory effects on the vasculature and immune response are involved in aneurysm formation. Taken together, these findings indicate that inflammation and the immune response play essential roles in the pathogenesis of IAs [6,26]. Another study also reported that immunoglobulins participate in the pathogenesis of aneurysms. The roles of immune response pathways in aneurysm rupture suggest that anti-inflammatory and immune-modifying drugs are interesting candidate therapeutics in the prevention of aneurysm rupture [27]. Matrix Metalloproteinases (MMPs) [28], a family of extracellular matrix-degrading collagenases secreted by macrophages, are crucial proteins in aneurysmal inflammation. Inflammation characterized by the release of MMPs by macrophages with degradation of the vascular extracellular matrix is believed to be shared by several vascular diseases, such as atherosclerosis and abdominal aortic aneurysms [29]. A rat model showed that macrophage-derived extracellular matrix remodeling was partially mediated by MMP2 and MMP9 induction [30]. In addition, studies have shown that the levels of MCP1, a macrophageattracting cytokine, in humans and animal models of IA are positively related to IA formation. MCP1- knockout mice showed reduced IA formation, inhibited macrophage aggregation, and downregulated expression of MMP2 and MMP9 [31]. Patients with more than one aneurysm display higher levels of MCP1 than patients with just one aneurysm [25]. Recently, Hasan et al. investigated the role of mast cells and macrophage imbalance in human tissue samples and found that inflammatory cell invasion is also associated with IA rupture [32]. They found that macrophages in atherosclerotic plaques can fall into two main subtypes: CD14 high/CD16 low and CD14 low/CD16 high. These two subtypes play opposite roles in inflammatory processes [33]. However, some studies have pointed out that immunostaining of surgically resected aneurysmal dome tissue reveals that proinflammatory and anti-inflammatory subset macrophages are in balance with a few mast cells present in unruptured IA walls [34]. These studies also indicated a deviation from the critical macrophage balance in ruptured aneurysm walls, which is a considerable excess of pro-inflammatory macrophage expression over anti-inflammatory macrophage expression in that situation. Leukocyte invasion is commonly observed during the progression of IAs. A recent study also detailed the patientspecific expression levels of two other cytokines, IL1β and TNFα. All three regulatory cytokines are expressed at increased levels in IAs compared with controls [31]. Furthermore, the immune response and chronic inflammation are important factors leading to atherosclerosis [3], which has been deemed a contributor to cerebral aneurysm formation and progression, because individuals with IAs and atherosclerosis share common risk factors, such as smoking and arterial hypertension. These immunologic responses suggest that inflammatory mediators are linked to the formation, progression, and rupture of cerebral aneurysms [3].
Conclusion
Our bioinformatic analysis identified 829 DEGs that might play crucial roles in the occurrence and development of IAs. TNF, as one of the hub genes, has an important role in the development of IAs. Thus, TNF can be used as a candidate gene to predict the occurrence of IAs. The immune response and inflammation play important roles in the progression of IAs. We provided a direction for the diagnosis and genomic individualized treatment of IAs.
Fund Resource
This study was approval by National Natural Science Foundation of China (81971870).
References
- Boissonneau S, Graillon T, Meyer M, Brunel H, Fuentes S, Dufour H. Intracranial Giant Mycotic Aneurysm without Endocarditis and Vasculitis: Report of Rare Entity and Review of Literature. World Neurosurg. 2018; 119: 353-357.
- Etminan N, Rinkel GJ. Unruptured intracranial aneurysms: development, rupture and preventive management. Nat Rev Neurol. 2017; 13: 126.
- Shimizu K, Kushamae M, Mizutani T, Aoki T. Intracranial aneurysm as a macrophage-mediated inflammatory disease. Neurologia medico-chirurgica. 2019; 59: 126-132.
- Wang C, Qu B, Wang Z, Ju J, Wang Y, Wang Z, et al. Proteomic identification of differentially expressed proteins in vascular wall of patients with ruptured intracranial aneurysms. Atherosclerosis. 2015; 238: 201-206.
- Jiang P, Wu J, Chen X, Ning B, Liu Q, Li Z, et al. Quantitative proteomics analysis of differentially expressed proteins in ruptured and unruptured cerebral aneurysms by iTRAQ. Journal of Proteomics 2018; 182: 45-52.
- Krischek B, Kasuya H, Tajima A, Akagawa H, Sasaki T, Yoneyama T, et al. Network-based gene expression analysis of intracranial aneurysm tissue reveals role of antigen presenting cells. Neuroscience. 2008; 154: 1398-1407.
- Wei L, Wang Q, Zhang Y, Yang C, Guan H, Chen Y, et al. Identification of key genes, transcription factors and microRNAs involved in intracranial aneurysm. Molecular medicine reports. 2018; 17: 891-897.
- Djelilovic-Vranic J, Basic-Kes V, Tiric-Campara M, Djozic E, Kulenovic J. Follow-up of vasospasm by Transcranial Doppler Sonography (TCD) in Subarachnoid Hemorrhage (SAH). Acta Informatica Medica. 2017; 25: 14-18.
- Macdonald RL. Origins of the concept of vasospasm. Stroke. 2016; 47: e11-e15.
- Liu H-J, Zhou H, Lu D-L, Jiao Y-B, Chen S-F, Cheng J, et al. Intracranial Mirror Aneurysm: Epidemiology, Rupture Risk, New Imaging, Controversies, and Treatment Strategies. World neurosurgery. 2019; 127: 165-175.
- Juvela S, Korja M. Intracranial aneurysm parameters for predicting a future subarachnoid hemorrhage: a long-term follow-up study. Neurosurgery. 2017; 81: 432-440.
- Okada T, Suzuki H. Toll-like receptor 4 as a possible therapeutic target for delayed brain injuries after aneurysmal subarachnoid hemorrhage. Neural regeneration research. 2017; 12: 193-196.
- Fan J, Yu L, Zhao J. Comparative transcriptome analysis reveals involvement of TLR-2 signaling in the pathogenesis of intracranial aneurysm. Journal of Clinical Neuroscience. 2018; 47: 258-263.
- Xiong X-Y, Liu L, Wang F-X, Yang Y-R, Hao J-W, Wang P-F, et al. Tolllike receptor 4/MyD88–mediated signaling of hepcidin expression causing brain iron accumulation, oxidative injury, and cognitive impairment after intracerebral hemorrhage. Circulation. 2016; 134: 1025-1038.
- Fontanella M, Rainero I, Gallone S, Rubino E, Fornaro R, Fenoglio P, et al. Interleukin-1 cluster gene polymorphisms and aneurysmal subarachnoid hemorrhage. Neurosurgery. 2010; 66: 1058-1063.
- Chalouhi N, Points L, Pierce GL, Ballas Z, Jabbour P, Hasan D. Localized increase of chemokines in the lumen of human cerebral aneurysms. Stroke. 2013; 44: 2594-2597.
- Kaminska J, Lyson T, Chrzanowski R, Sawicki K, Milewska AJ, Tylicka M, et al. Ratio of IL-8 in CSF versus Serum Is Elevated in Patients with Unruptured Brain Aneurysm. Journal of Clinical Medicine. 2020; 9: 1761.
- Lai X-L, Deng Z-F, Zhu X-G, Chen Z-H. Apc gene suppresses intracranial aneurysm formation and rupture through inhibiting the NF-κB signaling pathway mediated inflammatory response. Bioscience reports. 2019; 39.
- Ali MS, Starke RM, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, et al. TNF-α induces phenotypic modulation in cerebral vascular smooth muscle cells: implications for cerebral aneurysm pathology. Journal of Cerebral Blood Flow & Metabolism. 2013; 33: 1564-1573.
- Hu J, Luo J, Wang H, Wang C, Sun X, Li A, et al. Association of TNF-α- 3959T/C gene polymorphisms in the Chinese population with intracranial aneurysms. Journal of Molecular Neuroscience. 2017; 63: 349-354.
- Starke RM, Chalouhi N, Ali MS, Jabbour PM, Tjoumakaris SI, Gonzalez LF, et al. The role of oxidative stress in cerebral aneurysm formation and rupture. Current neurovascular research. 2013; 10: 247-255.
- Torres RD, Mancha F, Bustamante A, Canhao P, Fragata I, Montaner J. Usefulness of TNFR1 as biomarker of intracranial aneurysm in patients with spontaneous subarachnoid hemorrhage. Future Science OA. 2019; 6: FSO431.
- Fan W, Liu Y, Li C, Qu X, Zheng G, Zhang Q, et al. microRNA-331-3p maintains the contractile type of vascular smooth muscle cells by regulating TNF-α and CD14 in intracranial aneurysm. Neuropharmacology 2020; 164: 107858.
- Batra R, Suh MK, Carson JS, Dale MA, Meisinger TM, Fitzgerald M, et al. IL-1β (Interleukin-1β) and TNF-α (Tumor Necrosis Factor-α) Impact Abdominal Aortic Aneurysm Formation by Differential Effects on Macrophage Polarization. Arterioscler Thromb Vasc Biol. 2018; 38: 457-463.
- Zhang H-F, Zhao M-G, Liang G-B, Song Z-Q, Li Z-Q. Expression of proinflammatory cytokines and the risk of intracranial aneurysm. Inflammation. 2013; 36: 1195-1200.
- Chalouhi N, Ali MS, Starke RM, Jabbour PM, Tjoumakaris SI, Gonzalez LF, et al. Cigarette smoke and inflammation: role in cerebral aneurysm formation and rupture. Mediators of inflammation. 2012; 2012: 271582.
- Kleinloog R, Verweij BH, van der Vlies P, Deelen P, Swertz MA, de Muynck L, et al. RNA Sequencing Analysis of Intracranial Aneurysm Walls Reveals Involvement of Lysosomes and Immunoglobulins in Rupture. Stroke. 2016; 47: 1286-1293.
- Signorelli F, Sela S, Gesualdo L, Chevrel S, Tollet F, Pailler-Mattei C, et al. Hemodynamic Stress, Inflammation, and Intracranial Aneurysm Development and Rupture: A Systematic Review. World Neurosurg. 2018; 115: 234-244.
- Freestone T, Turner RJ, Coady A, Higman DJ, Greenhalgh RM, Powell JT. Inflammation and matrix metalloproteinases in the enlarging abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 1995; 15: 1145-1151.
- Wu R, Xu X, Liu A, Li W, Peng T, Qian Z, et al. [Pathophysiological roles of matrix metalloproteinases and nitric oxide synthase in cerebral aneurysm]. Zhonghua Yi Xue Za Zhi. 2014; 94: 2754-2756.
- Hudson JS, Hoyne DS, Hasan DM. Inflammation and human cerebral aneurysms: current and future treatment prospects. Future Neurol. 2013; 8: 10.
- Hasan D, Chalouhi N, Jabbour P, Hashimoto T. Macrophage imbalance (M1 vs. M2) and upregulation of mast cells in wall of ruptured human cerebral aneurysms: preliminary results. J Neuroinflammation. 2012; 9: 222.
- Gui T, Shimokado A, Sun Y, Akasaka T, Muragaki Y. Diverse roles of macrophages in atherosclerosis: from inflammatory biology to biomarker discovery. Mediators Inflamm. 2012; 2012: 693083.
- Hosaka K, Hoh BL. Inflammation and cerebral aneurysms. Transl Stroke Res. 2014; 5: 190-198.