
Review Article
Austin J Biomed Eng. 2014;1(5): 1001.
Trophic Factor Production by Glial Cells in the Treatment of Amyotrophic Lateral Sclerosis
Dennys CN, Franco MC and Estévez AG*
Department of Biomedical Sciences, University of Central Florida, USA
*Corresponding author: :Estévez AG, Burnett School of Biomedical Science, University of Central Florida, 6900 Lake Nona Blvd, Orlando, FL 32827, USA.
Received: August 10, 2014; Accepted: September 08, 2014; Published: September 10, 2014
Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease with unknown pathogenic causes. The identification of mutated genes in familial forms of ALS has lead to the development of animal models to study the mechanisms of motor neuron death. Transgenic animal models and human patients show that glutamate excitotoxicity, oxidative stress, protein aggregation and inflammation are hallmarks of the disease. Here we discuss the role of activated glial cells, specifically astrocytes and microglia on the disease pathogenesis and progression. Additionally, we discuss the current evidence showing that the induction of trophic factor production by glial cells may be an effective therapeutic strategy for the treatment of ALS.
Keywords: Amyotrophic lateral sclerosis; Astrocytes; Microglia; Trophic factors
Abbreviations
ALS: Amyotrophic Lateral Sclerosis; SOD: Superoxide Dismutase; GLT-1: Glutamate Transporter-1; EAAT2: Excitatory Amino Acid Transporter; AMPA: α-Amino-3-Hydroxy-5-Methyl- 4-Isoxazolepropionic Acid; Hsp90: Heat Shock Protein 90; Nrf2: Nuclear Factor Like 2; NOX: NADPH Oxidase; MCP1: Monocyte Chemo Attractant Protein; CCL5: Chemokine (CC motif) Ligand 5; TNF: Tumor Necrosis Factor; IL-1A: Interleukin-1A; IL-1B: Interleukin-1B; IL-1RA: Interleukin-1RA; M-CSF: Macrophage Colony Stimulating Factor; COX-2: Cyclooxygenase-2; iNOS: inducible nitric oxide synthase; Arg1: arginase-1; BDNF: brain derived neurotrophic factor; GDNF: Glial Derived Neurotrophic Factor; CNTF: Ciliaryneurotrophic Factor; VEGF: Vascular Endothelial Growth Factor; IGF1: Insulin Growth Factor-1; Hsp70: Heat Shock Protein 70
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease affecting 2 people every 100:000 populations. Most of the affected individuals live an average of 3-5 years after diagnosis: but some individuals live longer. Approximately 10% of cases are hereditary or familial and are referred as familial ALS. The other 90% of ALS cases are sporadic: for which the causes of the disease remain unknown. The identification of genes which mutations are associated to ALS allowed the development of transgenic mouse models of the disease. The discovery that 20-30% of the familial cases of ALS were linked to mutations in the gene of the antioxidant enzyme superoxide dismutase (SOD) lead to the development of a variety of transgenic mouse models that develop a disease with the general characteristics and symptoms of ALS[1-10]. In spite of the initial enthusiasm that the transgenic model would provide a rapid understanding of the disease leading to a cure: 20 years later there are no new treatments for ALS. The transgenic mouse models have become under attack as a tool to study the disease. Transgenic ALS mouse models over expressing mutant SOD not only have failed as a tool to help elucidate the mechanism of SOD toxicity: but also failed to be predictive for the development of successful human therapies.
However: transgenic animals recapitulate many of the characteristics of the disease and have become useful tools to elucidate the role of other cell types in the pathogenesis of ALS. It is now widely accepted that motor neurons are not the only cell type affected in ALS. During symptom onset and as disease progresses there is an increase in activated astrocytes and microglia [11,12]. These cells are also activated in the spinal cord of post mortem tissues of ALS patients [13-17]. Recently many studies have focused on the contribution of glial cells to disease onset and progression.
Amyotrophic Lateral Sclerosis
A number of different hypotheses have been developed over the years to explain motor neuron degeneration in ALS, including glutamate excitotoxicity, oxidative stress: protein aggregation and inflammation (Figure 1) [18]. The first hypothesis to explain the pathology of ALS was glutamate excitotoxicity [19]. In the G93A SOD mouse and rat ALS models, the levels and activity of glial glutamate transporter (GLT-1) were reduced [20-23]. Shortly after, a reduction in the expression of the human equivalent of GLT-1: EAAT-2, was observed in the motor cortex and spinal cord of ALS patients [24,25]. In addition, glutamate levels in the cerebral-spinal fluid were increased in ALS patients [26,27]. The decrease of EAAT- 2expressionwas proposed as the cause of extracellular glutamate accumulation: which in turn stimulated motor neuron degeneration through hyper activation of the AMPA glutamate receptors [28]. A similar decrease in glutamate transporter activity and increased levels of glutamate is present in the cerebral spinal fluid and plasma of mutant SOD mouse models [7,21,22,29]. Suggesting that the selective loss of glial GLT-1 causes the increase in glutamate levels both in vivo and in vitro [30]. Further support comes from delayed motor neuron degeneration in G93A mutant SOD transgenic models of ALS over expressing GLT-1[31], suggesting that restoration of glutamate uptake can delay disease progression. These observations were among the first to imply that glial cell dysfunction contributes to the progressive loss of motor neurons in ALS.
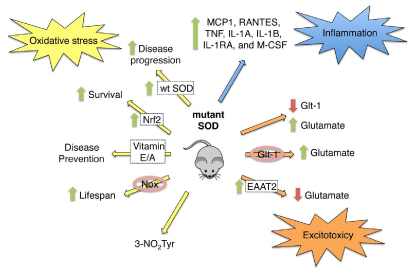
Figure 1: Role of oxidative stress, inflammation and excitoxicity in ALS.
As mentioned before, the first identified genetic cause of ALS was mutations in the gene of SOD [1]. At present more than 100 point and deletion mutations in the SOD gene have been identified [32,33]. In vitro and in vivo evidence support that SOD toxicity is consequence of a gain-in-function and not due to loss of enzymatic function [34,35]. Genetic deletion of SOD in transgenic mice does not induce motor neuron death [36]. Over expression of wild type human SOD does not protect against mutant SOD toxicity, in fact over expression of wild type SOD has no effect [37] or accelerates the death of the animals [38-40]. These results support that a gain in toxic function is responsible for the role of mutant SOD in the pathogenesis of ALS. However, 20 years after the discovery those mutations in SOD were the cause of ALS, the mechanism by which mutant SOD stimulates motor neuron death is still unknown and highly controversial.
Other common feature in familial and sporadic patients as well as in animal models of ALS is the presence of markers for the production of reactive oxygen and nitrogen species. The formation of peroxynitrite: the product of the diffusion limited reaction of nitric oxide and superoxide [41] has been proposed to participate in the pathogenesis of ALS [42,43]. Nitro tyrosine, a peroxynitrite foot print is present in glial cells and motor neurons from postmortem spinal cords of ALS patients and transgenic mutant SOD mouse [8, 44- 57]. One hypothesis of mutant SOD toxicity is based on the mutant enzyme’s lower affinity for zinc [58,59]. Zinc deficient SOD triggers superoxide- and nitric oxide-dependent motor neuron death [60]. Interestingly: wild type SOD enhances zinc-deficient SOD stimulated motor neuron death by stabilizing mutant SOD and preventing its aggregation [61]. In addition, the mutant enzyme catalyzes protein nitration by peroxynitrite [62]. Tyrosine nitration precedes motor neuron death induced by zinc-deficient SOD [60], suggesting that formation of nitro tyrosine could mediate the induction of cell death. Nitration of tyrosine residues has been linked to induction of cell death for over 20 years. Tyrosine-containing peptides indeed prevent peroxynitrite-induced apoptosis in PC12 cells: demonstrating that tyrosine nitration is the actual cause of cell death [63]. Nitration of the heat shock protein 90 (Hsp90) on a single tyrosine residue turns the pro-survival chaperone into a toxic protein. This toxic nitrated Hsp90 is present in motor neurons of ALS patients and animal models of ALS [64]. In aggregate: these results represent the first integrated hypothesis explaining the toxicity of mutant SOD as a catalyst of Hsp90 nitration: supporting nitrated Hsp90 toxic activity in motor neurons. Further investigation is necessary to mechanistically link mutant SOD with Hsp90 nitration.
Further support for the participation of oxidative stress in the pathogenesis of ALS is provided by studies that crossbreed the G93A SOD transgenic mouse model of ALS with animals over expressing nuclear factor-like 2 (Nrf2): which enhances the Nrf2 mediated antioxidant response in astrocytes. Astrocytes Nrf2 over expression in transgenic mouse models of ALS increases survival, delays symptom onset and slows progression of the disease [65]. Gene deletion or pharmacological inhibition of the superoxide-producing enzyme NADPH Oxidase (NOX) also prolongs lifespan in mouse models of ALS [66-68]. In addition: antioxidant vitamins such as vitamin E have proved effective in animal models of ALS [69].
In summary, glutamate toxicity and oxidative stress contribute to the pathogenesis of ALS. Each one of these processes have glial cells as possible mediators of the toxic effects, suggesting that dysregulation of glial cells and neuro inflammation cause motor neuron loss of function and death in ALS.
Role of Glial Cells in ALS Pathology
Neuro inflammation, characterized by activated microglia and astrocytes is present in the spinal cord of patients and animal models of ALS (Figure 2) [11,70-73]. In addition to the morphological and molecular markers of microglia activation, immune cell recruitment is detected both in patients and animal models of ALS. Monocyte chemo attractant protein (MCP1) and chemokine ligands 5 (CCL5), markers of immune cell recruitment are present in cerebrospinal fluid and sera of ALS patient [70,74]. A similar increase in MCP1 is present in animal models of ALS [75]. Up regulation of proinflammatory cytokines and chemokines such as tumor necrosis factor (TNF), interleukin 1A (IL-1A), interleukin 1B (IL-1B), interleukin-1RA (IL- 1RA), and macrophage colony stimulating factor (M-CSF) is also found in patients and animal models of ALS [76-80]. There is also an increase in cyclooxygenases 2 (COX-2) and prostaglandin E2 in both ALS patients and mutant SOD mouse models [81-83]. In addition: a correlation between inducible nitric oxide synthase (iNOS) and arginase 1 (Arg1) positive microglia, markers of microglia activation and disease progression: is evident in mutant SOD mouse models of ALS [84]. Gene reduction of mutant SOD expression in microglia and macrophages slows disease progression without affecting disease onset [85]. Together these results suggest that microglia play a secondary role in disease progression after ALS is trigger by a different mechanism.
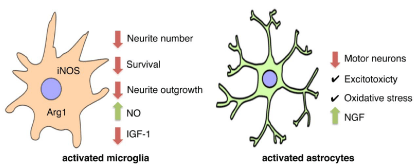
Figure 2: Roles of glial cells in ALS.
Activated astrocytes are found also in the spinal cord of both patients and mouse models of ALS [6,11,86-89]. Mutant SOD containing astrocytes are toxic to motor neurons in co-cultures [89- 95]. Similarly: astrocytes activated by different stimuli not only do not support motor neuron survival, but stimulate motor neuron death [89,96-99]. Growing evidence indicates that expression of mutant SOD in astrocytes is involved in disease progression, but does not affect the onset [87]. Activated astrocytes secrete nerve growth factor [100] at levels that are toxic to motor neuron [94,98,101-103]. These results suggest that astrocytes are an integral part of the pathogenic mechanism responsible for the progression of ALS.
In summary, glial cell dysfunction has been documented in animal and cell culture models of ALS as well as in ALS patients. Astrocytes and microglial have been shown to become activated and secrete toxic factors that lead to motor neuron death [92-95,99,104-106]. The disruption of normal glial cell function and trophic factor support suggests that targeting glial cells may be a therapeutic approach for the treatment of ALS.
Targeting Glial Cells in the Treatment of ALS
The observation that glial cells are activated upon symptom onset with the concomitant release of factors toxic to motor neurons, suggests that these cells may be potential therapeutic targets. Stimulating these cells to produce and secrete trophic factors that promote motor neuron survival may prove to be an effective strategy in the treatment of ALS. Riluzole is the only FDA approved drug for the treatment of ALS. However, the primary mechanism of action of Riluzole remains unknown. Riluzole has been shown to stimulate glutamate reuptake and to block sodium channels [107-112]. However, many other anti-glutamate agents have had no effect on patient survival, suggesting this is not the main protection mechanism. This lead to the hypothesis that Riluzole may be protective not only as an anti-excitotoxic agent but also by stimulating or inhibiting other cellular pathways [113-115]. Riluzole has also been shown to promote Trophic factor production in astrocytes [116,117], enhancing the protection provided by astrocytes to Trophic factor-deprived motor neurons. Exposing the conditioned media from astrocytes to heat or trypsin reverses this effect: which strongly suggest that Riluzole stimulates the production and release of Trophic factors [117]. Further studies showed that riluzole stimulates the expression of brain derived neurotrophic factor (BDNF) and glial derived neurotrophic factor (GDNF)in cortical astrocytes [116]. These results suggest that the effectiveness of riluzole may be due to an increase in trophic factor production by glial cells.
Riluzole only extends patient lifespan from three months to a year [118-123]. Therefore a more effective therapeutic strategy must be developed. The type of trophic factor used for ALS treatment is critical as trophic factors have different effects depending on the mode of administration and their pharmacokinetic limitations and side effects [124]. BDNF has been tested in animal models of axotomy, excitotoxic legions and wobblers mice. There is an increase in motor neuron survival following gel foam and spinal cord injections, as well as after viral delivery of BDNF to the animals [125-130]. However: there has been no benefit to BDNF treatments in human trials [131,132]. This might be due to inefficient crossing of BDNF through the blood brain barrier [124,133]. GDNF increases survival of mutant SOD motor neurons in vitro. In transgenic mice: viral delivery or muscle derived GDNF expression also extends survival [125,128,134- 136]. In contrast, the expression of GDNF in the central nervous tissue has no effect on motor neuron survival [135,137]. There is already an increase in GDNF levels in the cerebral spinal fluid of ALS patients [138], and this may explain why additional GDNF in the central nervous system has no effect in animal models. GDNF has not been tested in human trials due to the controversial evidence on treatment effectiveness. GDNF is also unable to cross the blood brain barrier, requiring more invasive drug delivery methods [139]. Finally, ciliaryneurotrophic factor(CNTF) has been shown to increase motor neuron survival after axotomy in a rat model [140]. However, no benefits were observed in human trials and the extreme side effects lead to termination of treatment in many cases [141].
Nontraditional neurotrophic factors have also been investigated for the treatment of ALS. Vascular endothelial growth factor (VEGF) is a neuro protective factor that is produced by astrocytes and Schwann cells [142]. Delivery of VEGF using viral vectors improves motor neuron function as well as numbers [143]. Intravenous injections of VEGF also improve motor neuron function, increase number of motor neurons and neuromuscular junctions and improve survival in animal models of ALS [144,145]. In addition, transplantation of human neural stem cells overexpressing VEGF in animal models of the disease improves motor neuron function as well as survival [146]. Finally: over expression of VEGF in neurons from ALS transgenic animals improves motor neuron survival and preserves neuromuscular junctions by reducing astrogliosis [147,148]. VEGF is down regulated in ALS patients [149,150], and there is currently a clinical trial under way to investigate VEGF as a therapeutic target for ALS patients. Insulin growth factor 1 (IGF1) is another neuro protective factor under investigation. IGF1 is produced and secreted by astrocytes, oligodendricytes, microglia and Schwann cells. Viral and intrathecal delivery of IGF1 at various stages of disease progression results in improved function and survival of motor neurons in transgenic mouse models of ALS. Muscle specific expression of IGF1 also improves motor neuron function and survival [151-156]. Human trials utilizing IGF1 treatment have shown a slower decline of motor neuron function where as one other has had no effect [157-160].
The induction of the heat shock response can also be neuro protective. Both muscle and astrocytes secrete heat shock protein 70 (Hsp70) under normal conditions. However, over expression of Hsp70 within the spinal cord, but not the muscle, can protect motor neurons from program cell death during development [161]. The observation that only centrally derived Hsp70 is neuro protective may suggest that the cellular source of a trophic factor is important. This may explain why clinical trials to date have been unsuccessful.
Activation of astrocytes and microglia in ALS results in the secretion of toxic factors that induce motor neuron death, which enhances disease progression. Riluzole stimulates astrocytes to produce trophic factors that prevent motor neuron death following trophic factor deprivation, and may explain the extension of animal survival in mouse models of ALS. Over expression of neurotrophic factors is protective in animal models of ALS. However these tropic factor therapies are not effective in human patients. [69,162-165]. The particular trophic factor under study may have difficulties reaching the target cell type at therapeutically relevant concentrations. In fact, it may not be able to cross the blood brain barrier [124]. The Trophic factors currently under investigation may not be the most potent factors available to improve motor neuron survival. Successful clinical trials may require the right Trophic factor being produced by the right cell type to be effective. Further study in this area is needed to address these issues.
Conclusion
The development of ALS depends on a variety of factors that contribute to disease pathology. The role of inflammation and oxidative stress points towards glial cell dysfunction during disease progression. Secretion of toxic factors by these cell types is associated with neuronal death. Stimulation of pro-survival neurotrophic factor production by these cell types has been shown to prevent motor neuron death. Recombinant protein therapies have in some cases had a small therapeutic effect in humans. An alternative to these therapies would be to target glial cells to secrete trophic factors locally to ensure the correct therapeutic level of these factors reaching the motor neurons. Therefore, future studies should investigate the induction of trophic factor production in vivo to treat ALS. In addition, development of in vitro culture and co-culture of glial cells with motor neurons and muscle humanized platforms for high throughput screening of agents with protective activities could accelerate the discovery of new therapeutic agents for ALS.
Acknowledgement
This work was supported by the Burnett School of Biomedical Sciences, College of Medicine, University of Central Florida and the National Institute of Health (NIH Grant NS36761 to A.G.E.).
References
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993; 362: 59-62.
- Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993; 261: 1047-1051.
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994; 264: 1772-1775.
- Wang L, Sharma K, Deng HX, Siddique T, Grisotti G, Liu E, et al. Restricted expression of mutant SOD1 in spinal motor neurons and interneurons induces motor neuron pathology. Neurobiol Dis. 2008; 29: 400-408.
- Wang L, Deng HX, Grisotti G, Zhai H, Siddique T, Roos RP. Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Hum Mol Genet. 2009; 18: 1642-1651.
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995; 14: 1105-1116.
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997; 18: 327-338.
- Bruijn LI, Beal MF, Becher MW, Schulz JB, Wong PC, Price DL, et al. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci U S A. 1997; 94: 7606-7611.
- Wong PC, Cai H, Borchelt DR, Price DL. Genetically engineered mouse models of neurodegenerative diseases. Nat Neurosci. 2002; 5: 633-639.
- Martin LJ, Price AC, Kaiser A, Shaikh AY, Liu Z. Mechanisms for neuronal degeneration in amyotrophic lateral sclerosis and in models of motor neuron death (Review). Int J Mol Med. 2000; 5: 3-13.
- Barbeito L, Pehar M, Cassina P, Vargas MR, Peluffo H, Viera L, et al. Role of astroglia in the pathogenesis of amyotrophic lateral sclerosis. Brain Res Rev. 2004; 47: 263-274.
- Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998; 23: 249-256.
- Schiffer D, Cordera S, Cavalla P, Migheli A. Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci. 1996; 139 Suppl: 27-33.
- Jossan SS, Ekblom J, Aquilonius SM, Oreland L. Monoamine oxidase-B in motor cortex and spinal cord in amyotrophic lateral sclerosis studied by quantitative autoradiography. J Neural Transm Suppl. 1994; 41: 243-248.
- Banati RB, Gehrmann J, Kellner M, Holsboer F. Antibodies against microglia/brain macrophages in the cerebrospinal fluid of a patient with acute amyotrophic lateral sclerosis and presenile dementia. Clin Neuropathol. 1995; 14: 197-200.
- Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992; 140: 691-707.
- Engelhardt JI, Appel SH. IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch Neurol. 1990; 47: 1210-1216.
- Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009; 65 Suppl 1: S3-9.
- Rothstein JD. Excitotoxicity hypothesis. Neurology. 1996; 47: S19-25.
- Trotti D, Rolfs A, Danbolt NC, Brown RH Jr, Hediger MA. SOD 1 mutants linked to amyotrophic lateral sclerosis selectivity inactivate a glial glutamate transporter. Nature Neurosci. 1999; 2: 427-433.
- Canton T, Pratt J, Stutzmann JM, Imperato A, Boireau A. Glutamate uptake is decreased tardively in the spinal cord of FALS mice. Neuroreport. 1998; 9: 775-778.
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA. 2002; 99: 1604-1609.
- Bendotti C, Tortarolo M, Suchak SK, Calvaresi N, Carvelli L, Bastone A, et al. Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. Journal of Neurochemsitry. 2001; 79: 737-746.
- Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992; 326: 1464-1468.
- Fray AE, Ince PG, Banner SJ, Milton ID, Usher PA, Cookson MR, et al. The expression of the glial glutamate transporter protein EAAT2 in motor neuron disease: an immunohistochemical study. Eur J Neurosci. 1998; 10: 2481-2489.
- Plaitakis A, Constantakakis E, Smith J. The neuroexcitotoxic amino acids glutamate and aspartate are altered in the spinal cord and brain in amyotrophic lateral sclerosis. Ann Neurol. 1988; 24: 446-449.
- Plaitakis A. Glutamate dysfunction and selective motor neuron degeneration in amyotrophic lateral sclerosis: a hypothesis. Ann Neurol. 1990; 28: 3-8.
- Couratier P, Hugon J, Sindou P, Vallat JM, Dumas M. Cell culture evidence for neuronal degeneration in amyotrophic lateral sclerosis being linked to glutamate AMPA/kainate receptors. Lancet. 1993; 341: 265-268.
- Shaw PJ, Forrest V, Ince PG, Richardson JP, Wastell HJ. CSF and plasma amino acid levels in motor neuron disease: elevation of CSF glutamate in a subset of patients. Neurodegeneration. 1995; 4: 209-216.
- Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci U S A. 1993; 90: 6591-6595.
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuro protection by increasing glutamate transporter expression. Nature. 2005; 433: 73-77.
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013; 14: 248-264.
- Franco MC, et al. Superoxide Dismutase and Oxidative Stress in Amyotrophic Lateral Sclerosis. Current Advances in Amyotrophic Lateral Sclerosis. Rejeka, Croatia: InThech. 2013.
- Borchelt DR, Lee MK, Slunt HS, Guarnieri M, Xu ZS, Wong PC, et al. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc Natl Acad Sci USA. 1994; 91: 8292-8296.
- Borchelt DR, Guarnieri M, Wong PC, Lee MK, Slunt HS, Xu ZS, et al. Superoxide dismutase 1 subunits with mutations linked to familial amyotrophic lateral sclerosis do not affect wild-type subunit function. J Biol Chem. 1995; 270: 3234-3238.
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996; 13: 43-47.
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, et al. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998; 281: 1851-1854.
- Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci USA. 2006; 103: 7142-7147.
- Furukawa Y, Fu R, Deng HX, Siddique T, O'Halloran TV. Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. PNAS. 2006; 103: 7148-7153.
- Wang H, Ying Z, Wang G. Ataxin-3 regulates aggresome formation of copper-zinc superoxide dismutase (SOD1) by editing K63-linked polyubiquitin chains. J Biol Chem. 2012; 287: 28576-28585.
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990; 87: 1620-1624.
- Beckman JS, Estévez AG, Crow JP, Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001; 24: S15-20.
- Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxynitrite. Nature. 1993; 364: 584.
- Beal MF, Ferrante RJ, Browne SE, Matthews RT, Kowall NW, Brown RH Jr. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997; 42: 644-654.
- Abe K, Pan LH, Watanabe M, Konno H, Kato T, Itoyama Y. Upregulation of protein-tyrosine nitration in the anterior horn cells of amyotrophic lateral sclerosis. Neurol Res. 1997; 19: 124-128.
- Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, et al. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997; 69: 2064-2074.
- Ferrante RJ, Shinobu LA, Schulz JB, Matthews RT, Thomas CE, Kowall NW, et al. Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann Neurol. 1997; 42: 326-334.
- Franco MC, Ye Y, Refakis CA, Feldman JL, Stokes AL, Basso M, et al. Nitration of Hsp90 induces cell death. Proc Natl Acad Sci U S A. 2013; 110: E1102-1111.
- Casoni F, Basso M, Massignan T, Gianazza E, Cheroni C, Salmona M, et al. Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: possible multifunctional role in the pathogenesis. J Biol Chem. 2005; 280: 16295-16304.
- Basso M, Samengo G, Nardo G, Massignan T, D'Alessandro G, Tartari S, et al. Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS One. 2009; 4: e8130.
- Chou SM, Wang HS, Komai K. Colocalization of NOS and SOD1 in neurofilament accumulation within motor neurons of amyotrophic lateral sclerosis: an immunohistochemical study. J Chem Neuroanat. 1996; 10: 249-258.
- Abe K, Pan LH, Watanabe M, Kato T, Itoyama Y. Induction of nitrotyrosine-like immunoreactivity in the lower motor neuron of amyotrophic lateral sclerosis. Neurosci Lett. 1995; 199: 152-154.
- Cha CI, Chung YH, Shin CM, Shin DH, Kim YS, Gurney ME,et al. Immunocytochemical study on the distribution of nitrotyrosine in the brain of the transgenic mice expressing a human Cu/Zn SOD mutation. Brain Res. 2000; 853: 156-161.
- Sasaki S, Shibata N, Komori T, Iwata M. iNOS and nitrotyrosine immunoreactivity in amyotrophic lateral sclerosis. Neurosci Lett. 2000; 291: 44-48.
- Sasaki S, Warita H, Abe K, Iwata M. Inducible nitric oxide synthase (iNOS) and nitrotyrosine immunoreactivity in the spinal cords of transgenic mice with a G93A mutant SOD1 gene. J Neuropathol Exp Neurol. 2001; 60: 839-846.
- Tohgi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C. Remarkable increase in cerebrospinal fluid 3-nitrotyrosine in patients with sporadic amyotrophic lateral sclerosis. Ann Neurol. 1999; 46: 129-131.
- Petri S, Kiaei M, Wille E, Calingasan NY, Flint Beal M. Loss of Fas ligand-function improves survival in G93A-transgenic ALS mice. J Neurol Sci. 2006; 251: 44-49.
- Lyons TJ, Liu H, Goto JJ, Nersissian A, Roe JA, Graden JA, et al. Mutations in copper-zinc superoxide dismutase that cause amyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of the protein. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93: 12240-12244.
- Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 1997; 69: 1936-1944.
- Estévez AG, Crow JP, Sampson JB, Reiter C, Zhuang Y, Richardson GJ, et al. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999; 286: 2498-2500.
- Sahawneh MA, Ricart KC, Roberts BR, Bomben VC, Basso M, Ye Y, et al. Cu,Zn-superoxide dismutase increases toxicity of mutant and zinc-deficient superoxide dismutase by enhancing protein stability. J Biol Chem. 2010; 285: 33885-33897.
- Crow JP, Ye YZ, Strong M, Kirk M, Barnes S, Beckman JS. Superoxide dismutase catalyzes nitration of tyrosines by peroxynitrite in the rod and head domains of neurofilament-L. J Neurochem. 1997; 69: 1945-1953.
- Ye Y, Quijano C, Robinson KM, Ricart KC, Strayer AL, Sahawneh MA, et al. Prevention of peroxynitrite-induced apoptosis of motor neurons and PC12 cells by tyrosine-containing peptides. J Biol Chem. 2007; 282: 6324-6337.
- Franco MC, Ye Y, Refakis CA, Feldman JL, Stokes AL, Basso M, et al. Nitration of Hsp90 induces cell death. PNAS. 2013; 110: E1102-1111.
- Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008; 28: 13574-13581.
- Wu DC, Ré DB, Nagai M, Ischiropoulos H, Przedborski S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci USA. 2006; 103: 12132-12137.
- Marden JJ, Harraz MM, Williams AJ, Nelson K, Luo M, Paulson H, et al. Redox modifier genes in amyotrophic lateral sclerosis in mice. J Clin Invest. 2007; 117: 2913-2919.
- Boillée S, Cleveland DW. Revisiting oxidative damage in ALS: microglia, Nox, and mutant SOD1. J Clin Invest. 2008; 118: 474-478.
- Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, et al. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996; 39: 147-157.
- Henkel JS, Engelhardt JI, Siklós L, Simpson EP, Kim SH, Pan T, et al. Presence of dendritic cells, MCP-, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Annals of Neurology. 2004; 55: 221-235.
- McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve. 2002; 26: 459-470.
- Zhao W, Beers DR, Appel SH. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol. 2013; 8: 888-899.
- Evans MC, Couch Y, Sibson N, Turner MR. Inflammation and neurovascular changes in amyotrophic lateral sclerosis. Mol Cell Neurosci. 2013; 53: 34-41.
- Rentzos M, Nikolaou C, Rombos A, Boufidou F, Zoga M, Dimitrakopoulos A, et al. RANTES levels are elevated in serum and cerebrospinal fluid in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2007; 8: 283-287.
- Sargsyan SA, Blackburn DJ, Barber SC, Monk PN, Shaw PJ. Mutant SOD1 G93A microglia have an inflammatory phenotype and elevated production of MCP-1. Neuroreport. 2009; 20: 1450-1455.
- Kenneth Hensley RAF, Brian Gordon, Shenyun Mou, Quentin N Pye, Charles Stewart, Melinda West, et al. Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Journal of Neurochemistry. 2002; 82: 365-374.
- Hensley K, Fedynyshyn J, Ferrell S, Floyd RA, Gordon B, Grammas P, et al. Message and protein-level elevation of tumor necrosis factor a (TNFa) and TNFa-modulating cytokines in spinal cords of the G93A-SOD1 mouse model for amyotrophic lateral sclerosis. Neurobiology of Disease. 2003; 14: 74-80.
- Almer G, Guégan C, Teismann P, Naini A, Rosoklija G, Hays AP, et al. Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann Neurol. 2001; 49: 176-185.
- Yoshihara T, Ishigaki S, Yamamoto M, Liang Y, Niwa J, Takeuchi H, et al. Differential expression of inflammation- and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 2002; 80: 158-167.
- Gowing G, Lalancette-Hébert M, Audet JN, Dequen F, Julien JP. Macrophage colony stimulating factor (M-CSF) exacerbates ALS disease in a mouse model through altered responses of microglia expressing mutant superoxide dismutase. Exp Neurol. 2009; 220: 267-275.
- Christian Maihöfner SPC, Markus Bergmann, Winfried Neuhuber, Bernhard Neundörfer, Dieter Heuss. Expression and localization of cyclooxygenase-1 and -2 in human sporadic amyotrophic lateral sclerosis. European Journal of Neuroscience. 2003; 18: 1527-1534.
- Yasojima K, Tourtellotte WW, McGeer EG, McGeer PL. Marked increase in cyclooxygenase-2 in ALS spinal cord: implications for therapy. Neurology. 2001; 57: 952-956.
- Liang X, Wang Q, Shi J, Lokteva L, Breyer RM, Montine TJ, et al. The prostaglandin E2 EP2 receptor accelerates disease progression and inflammation in a model of amyotrophic lateral sclerosis. Ann Neurol. 2008; 64: 304-314.
- Katherine E Lewis, AL Rasmussen, William Bennett, Anna King, Adrian K West, Roger S Chung, et al. Microglia and motor neurons during disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis: changes in arginase1 and inducible nitric oxide synthase. Journal of Neuro inflammation. 2014; 11.
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006; 312: 1389-1392.
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006; 52: 39-59.
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008; 11: 251-253.
- Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci. 2007; 10: 1355-1360.
- Díaz-Amarilla P, Olivera-Bravo S, Trias E, Cragnolini A, Martínez-Palma L, Cassina P, et al. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108: 18126-18131.
- Pehar M, Vargas MR, Robinson KM, Cassina P, Díaz-Amarilla PJ, Hagen TM, et al. Mitochondrial Superoxide Production and Nuclear Factor Erythroid 2-Related Factor 2 Activation in p75 Neurotrophin Receptor-Induced Motor Neuron Apoptosis. J. Neurosci. 2007; 27: 7777-7785.
- Aebischer J, Cassina P, Otsmane B, Moumen A, Seilhean D, Meininger V, et al. IFNγ triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1. Cell Death Differ. 2011; 18: 754-768.
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007; 10: 615-622.
- Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007; 10: 608-614.
- Ferraiuolo L, Higginbottom A, Heath PR, Barber S, Greenald D, Kirby J, et al. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain. 2011; 134: 2627-2641.
- Basso M, Pozzi S, Tortarolo M, Fiordaliso F, Bisighini C, Pasetto L, et al. Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J Biol Chem. 2013; 288: 15699-15711.
- Cassina P, Peluffo H, Pehar M, Martinez-Palma L, Ressia A, Beckman JS, et al. Peroxynitrite triggers a phenotypic transformation in spinal cord astrocytes that induces motor neuron apoptosis. J Neurosci Res. 2002; 67: 21-29.
- Shin JT, Barbeito L, MacMillan-Crow LA, Beckman JS, Thompson JA. Acidic fibroblast growth factor enhances peroxynitrite-induced apoptosis in primary murine fibroblasts. Arch Biochem Biophys. 1996; 335: 32-41.
- Pehar M, Cassina P, Vargas MR, Castellanos R, Viera L, Beckman JS, et al. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2004; 89: 464-473.
- Cassina P, Pehar M, Vargas MR, Castellanos R, Barbeito AG, Estévez AG, et al. Astrocyte activation by fibroblast growth factor-1 and motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2005; 93: 38-46.
- Turner BJ, Cheah IK, Macfarlane KJ, Lopes EC, Petratos S, Langford SJ, et al. Antisense peptide nucleic acid-mediated knockdown of the p75 neurotrophin receptor delays motor neuron disease in mutant SOD1 transgenic mice. J Neurochem. 2003; 87: 752-763.
- Wiese S, Metzger F, Holtmann B, Sendtner M. The role of p75NTR in modulating neurotrophin survival effects in developing motoneurons. Eur J Neurosci. 1999; 11: 1668-1676.
- Ricart K, J Pearson R Jr, Viera L, Cassina P, Kamaid A, Carroll SL, et al. Interactions between beta-neuregulin and neurotrophins in motor neuron apoptosis. J Neurochem. 2006; 97: 222-233.
- Pehar M, Vargas MR, Robinson KM, Cassina P, England P, Beckman JS, et al. Peroxynitrite transforms nerve growth factor into an apoptotic factor for motor neurons. Free Radic Biol Med. 2006; 41: 1632-1644.
- Kunze A, Lengacher S, Dirren E, Aebischer P, Magistretti PJ, Renaud P. Astrocyte-neuron co-culture on microchips based on the model of SOD mutation to mimic ALS. Integr Biol. 2013; 5: 964-975.
- Xiao Q, Zhao W, Beers DR, Yen AA, Xie W, Henkel JS, et al. Mutant SOD1 (G93A) microglia are more neurotoxic relative to wild-type microglia. J Neurochem. 2007; 102: 2008-2019.
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006; 312: 1389-1392.
- Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008; 578: 171-176.
- Niebroj-Dobosz I, Janik P, KwieciÅ, ski H. Effect of Riluzole on serum amino acids in patients with amyotrophic lateral sclerosis. Acta Neurol Scand. 2002; 106: 39-43.
- Martin D, Thompson MA, Nadler JV. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol. 1993; 250: 473-476.
- Beltran-Parrazal L, Charles A. Riluzole inhibits spontaneous Ca2+ signaling in neuroendocrine cells by activation of K+ channels and inhibition of Na+ channels. Br J Pharmacol. 2003; 140: 881-888.
- Mohammadi B, Lang N, Dengler R, Bufler J. Interaction of high concentrations of riluzole with recombinant skeletal muscle sodium channels and adult-type nicotinic receptor channels. Muscle Nerve. 2002; 26: 539-545.
- Wang YJ, Lin MW, Lin AA, Wu SN. Riluzole-induced block of voltage-gated Na+ current and activation of BKCa channels in cultured differentiated human skeletal muscle cells. Life Sci. 2008; 82: 11-20.
- Meininger V, Lacomblez L, Salachas F. What has changed with riluzole? J Neurol. 2000; 247: 19-22.
- Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci Ther. 2011; 17: 4-31.
- Cheah BC, Vucic S, Krishnan AV, Kiernan MC. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Curr Med Chem. 2010; 17: 1942-1199.
- Mizuta I, Ohta M, Ohta K, Nishimura M, Mizuta E, Kuno S. Riluzole stimulates nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor synthesis in cultured mouse astrocytes. Neuroscience Letters. 2001; 310: 117-120.
- Peluffo H, Estevez A, Barbeito L, Stutzmann JM. Riluzole promotes survival of rat motoneurons in vitro by stimulating trophic activity produced by spinal astrocyte monolayers. Neuroscience Letters. 1997; 228: 207-211.
- Lacomblez L, et al. Long-term safety of riluzole in amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis and other motor neuron disorders: official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2002; 3: 23-29.
- Bensimon G, Lacomblez L, Delumeau JC, Bejuit R, Truffinet P, Meininger V. Riluzole/ALS Study Group II. A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. J Neurol. 2002; 249: 609-615.
- Riviere M, Meininger V, Zeisser P, Munsat T. An analysis of extended survival in patients with amyotrophic lateral sclerosis treated with riluzole. Arch Neurol. 1998; 55: 526-528.
- Meininger V, Dib M, Aubin F, Jourdain G, Zeisser P. The Riluzole Early Access Programme: descriptive analysis of 844 patients in France. ALS/Riluzole Study Group III. J Neurol. 1997; 244: S22-25.
- Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996; 347: 1425-1431.
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994; 330: 585-591.
- Gould TW, Oppenheim RW. Motor neuron trophic factors: therapeutic use in ALS? Brain Res Rev. 2011; 67: 1-39.
- Giménez y Ribotta M, Revah F, Pradier L, Loquet I, Mallet J, Privat A. Prevention of motoneuron death by adenovirus-mediated neurotrophic factors. J Neurosci Res. 1997; 48: 281-285.
- Bemelmans AP, Husson I, Jaquet M, Mallet J, Kosofsky BE, Gressens P. Lentiviral-mediated gene transfer of brain-derived neurotrophic factor is neuroprotective in a mouse model of neonatal excitotoxic challenge. J Neurosci Res. 2006; 83: 50-60.
- Ikeda K, Klinkosz B, Greene T, Cedarbaum JM, Wong V, Lindsay RM, et al. Effects of brain-derived neurotrophic factor on motor dysfunction in wobbler mouse motor neuron disease. Ann Neurol. 1995; 37: 505-511.
- Park S, Kim HT, Yun S, Kim IS, Lee J, Lee IS, et al. Growth factor-expressing human neural progenitor cell grafts protect motor neurons but do not ameliorate motor performance and survival in ALS mice. Exp Mol Med. 2009; 41: 487-500.
- Sendtner M, Holtmann B, Kolbeck R, Thoenen H, Barde YA. Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature. 1992; 360: 757-759.
- Yan Q, Elliott J, Snider WD. Brain-derived neurotrophic factor rescues spinal motor neurons from axotomy-induced cell death. Nature. 1992; 360: 753-755.
- Beck M, Flachenecker P, Magnus T, Giess R, Reiners K, Toyka KV, et al. Autonomic dysfunction in ALS: a preliminary study on the effects of intrathecal BDNF. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005; 6: 100-103.
- Kalra S, Genge A, Arnold DL. A prospective, randomized, placebo-controlled evaluation of corticoneuronal response to intrathecal BDNF therapy in ALS using magnetic resonance spectroscopy: feasibility and results. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003; 4: 22-26.
- Yan Q, Matheson C, Sun J, Radeke MJ, Feinstein SC, Miller JA. Distribution of intracerebral ventricularly administered neurotrophins in rat brain and its correlation with trk receptor expression. Exp Neurol. 1994; 127: 23-36.
- Suzuki M, McHugh J, Tork C, Shelley B, Klein SM, Aebischer P, et al. GDNF secreting human neural progenitor cells protect dying motor neurons, but not their projection to muscle, in a rat model of familial ALS. PLoS One. 2007; 2: e689.
- Li W, Brakefield D, Pan Y, Hunter D, Myckatyn TM, Parsadanian A. Muscle-derived but not centrally derived transgene GDNF is neuroprotective in G93A-SOD1 mouse model of ALS. Exp Neurol. 2007; 203: 457-471.
- Mohajeri MH, Figlewicz DA, Bohn MC. Intramuscular grafts of myoblasts genetically modified to secrete glial cell line-derived neurotrophic factor prevent motoneuron loss and disease progression in a mouse model of familial amyotrophic lateral sclerosis. Hum Gene Ther. 1999; 10:1853-1866.
- Acsadi G, Anguelov RA, Yang H, Toth G, Thomas R, Jani A, et al. Increased survival and function of SOD1 mice after glial cell-derived neurotrophic factor gene therapy. Hum Gene Ther. 2002; 13: 1047-1059.
- Grundström E, Lindholm D, Johansson A, Blennow K, Askmark H. GDNF but not BDNF is increased in cerebrospinal fluid in amyotrophic lateral sclerosis. Neuroreport. 2000; 11: 1781-1783.
- Kastin AJ, Akerstrom V, Pan W. Glial cell line-derived neurotrophic factor does not enter normal mouse brain. Neurosci Lett. 2003; 340: 239-241.
- Sendtner M, Kreutzberg GW, Thoenen H. Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature. 1990; 345: 440-441.
- ALS CNTF Treatment Study Group. A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis.. Neurology. 1996; 46: 1244-1249.
- Chow J, Ogunshola O, Fan SY, Li Y, Ment LR, Madri JA. Astrocyte-derived VEGF mediates survival and tube stabilization of hypoxic brain microvascular endothelial cells in vitro. Developmental Brain Research. 2001; 130: 123-132.
- Azzouz M, Ralph GS, Storkebaum E, Walmsley LE, Mitrophanous KA, Kingsman SM, et al. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature. 2004; 429: 413-417.
- Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, et al. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nature Neuroscience. 2005; 8: 85-92.
- Gupta R, Gray M, Chao T, Bear D, Modafferi E, Mozaffar T. Schwann cells up regulate vascular endothelial growth factor secondary to chronic nerve compression injury. Muscle Nerve. 2005; 31: 452-460.
- Hwang DH, Lee HJ, Park IH, Seok JI, Kim BG, Joo IS, et al. Intrathecal transplantation of human neural stem cells over expressing VEGF provide behavioral improvement, disease onset delay and survival extension in transgenic ALS mice. Gene Ther. 2009; 16: 1234-1244.
- Wang Y, Mao XO, Xie L, Banwait S, Marti HH, Greenberg DA, et al. Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J Neurosci. 2007; 27: 304-307.
- Zheng C, Sköld MK, Li J, Nennesmo I, Fadeel B, Henter JI. VEGF reduces astrogliosis and preserves neuromuscular junctions in ALS transgenic mice. Biochem Biophys Res Commun. 2007; 363: 989-993.
- IÅ‚zecka J. Cerebrospinal fluid vascular endothelial growth factor in patients with amyotrophic lateral sclerosis. Clin Neurol Neurosurg. 2004; 106: 289-293.
- Devos D, Moreau C, Lassalle P, Perez T, De Seze J, Brunaud-Danel V, et al. Low levels of the vascular endothelial growth factor in CSF from early ALS patients. Neurology. 2004; 62: 2127-2129.
- Franz CK, Federici T, Yang J, Backus C, Oh SS, Teng Q, et al. Intraspinal cord delivery of IGF-I mediated by adeno-associated virus 2 is neuroprotective in a rat model of familial ALS. Neurobiol Dis. 2009; 33: 473-481.
- Li L, Oppenheim RW, Lei M, Houenou LJ. Neurotrophic agents prevent motoneuron death following sciatic nerve section in the neonatal mouse. J Neurobiol. 1994; 25: 759-766.
- Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol. 2005; 168: 193-199.
- Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol. 2005; 168: 193-199.
- Lepore AC, Haenggeli C, Gasmi M, Bishop KM, Bartus RT, Maragakis NJ, et al. Intraparenchymal spinal cord delivery of adeno-associated virus IGF-1 is protective in the SOD1G93A model of ALS. Brain Res. 2007; 1185: 256-265.
- Messi ML, Clark HM, Prevette DM, Oppenheim RW, Delbono O. The lack of effect of specific overexpression of IGF-1 in the central nervous system or skeletal muscle on pathophysiology in the G93A SOD-1 mouse model of ALS. Exp Neurol. 2007; 207: 52-63.
- Lai EC FK, Festoff BW, Gawel MJ, Gelinas DF, Kratz R, Murphy MF, et al. Effect of recombinant human insulin-like growth factor-I on progression of ALS. A placebo-controlled study. The North America ALS/IGF-I Study Group. Neurology. 1997; 49: 1621-1630.
- Borasio GD, Robberecht W, Leigh PN, Emile J, Guiloff RJ, Jerusalem F, et al. A placebo-controlled trial of insulin-like growth factor-I in amyotrophic lateral sclerosis. European ALS/IGF-I Study Group. Neurology. 1998; 51: 583-586.
- Nagano I, Shiote M, Murakami T, Kamada H, Hamakawa Y, Matsubara E, et al. Beneficial effects of intrathecal IGF-1 administration in patients with amyotrophic lateral sclerosis. Neurol Res. 2005; 27: 768-772.
- Sorenson EJ, Windbank AJ, Mandrekar JN, Bamlet WR, Appel SH, Armon C, et al. Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology. 2008; 71: 1770-1775.
- Robinson MB, Tidwell JL, Gould T, Taylor AR, Newbern JM, Graves J, et al. Extracellular heat shock protein 70: a critical component for motoneuron survival. J Neurosci. 2005; 25: 9735-9745.
- Gurney ME, Fleck TJ, Himes CS, Hall ED. Riluzole preserves motor function in a transgenic model of familial amyotrophic lateral sclerosis. Neurology. 1998; 50: 62-66.
- Snow RJ, Turnbull J, da Silva S, Jiang F, Tarnopolsky MA. Creatine supplementation and riluzole treatment provide similar beneficial effects in copper, zinc superoxide dismutase (G93A) transgenic mice. Neuroscience. 2003; 119: 661-667.
- Waibel S, Reuter A, Malessa S, Blaugrund E, Ludolph AC. Rasagiline alone and in combination with riluzole prolongs survival in an ALS mouse model. J Neurol. 2004; 251: 1080-1084.
- Del Signore SJ, Amante DJ, Kim J, Stack EC, Goodrich S, Cormier K, et al. Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases. 2009; 10: 85-94.