
Abstract
Leber’s Hereditary Optic Neuropathy (LHON) is a maternally inherited mitochondrial disease, characterized by point mutations in mitochondrial DNA and subsequent mitochondrial dysfunction; which leads to a bilateral and painless loss of central vision. Unfortunately at this time, there is no proven treatment to prevent or reverse this optic neuropathy. Therefore, individualized low vision rehabilitation services and assistive devices are essential to help people with LHON regain their independence and quality of life. We highlight through a case study the importance of psychic support as well as the impact of low vision rehabilitation on a young man with LHON.
Keywords: Leber’s hereditary optic neuropathy; low vision rehabilitation.
Introduction
First reported by Von Graefe in 1858, then by Théodore Leber in 1871, Leber optic neuropathy is an inherited genetic disease of maternal transmission, characterised by the presence of mutations in the mitochondrial genome. Several mutations have been described, the main ones being 11778, 3460, 14484 and 15257; they affect genes coding for co-factors in the cellular respiratory chain. It is characterised by an acute or sub-acute, unilateral then rapidly bilateral decrease in profound visual acuity, mainly affecting young men.
Case Presentation
A 17-year-old patient presented with a progressive decline in visual acuity over the last year in the left eye and 5 months later in the right eye, accompanied by bothersome glare in daylight. Despite optical correction, the condition worsened. The medical history revealed no congenital or systemic diseases, no drug or substance abuse, and no family history of visual impairment. On presentation, the patient's Best Corrected Visual Acuity (BCVA) was 1/10 on the right and finger movement on the left. Following biomicroscopic assessment, the ocular surface, anterior segment and intraocular pressure were normal. Nonetheless, it was noted that a relative afferent pupillary deficit was present on the right side, alongside temporal sectorial papillary pallor in both eyes (Figure 1). There were no notable features found during neurological and general examinations. Fluorescein angiography revealed no abnormalities in the retention or leakage in the late phase, as illustrated in Figure 2. The visual field examination indicated a near-total deficiency in the left eye and a central scotoma in the right, with colour vision abnormalities evident along the red-green axis. In the EPI, the P2 wave amplitudes were significantly low in response to all eye stimuli (refer to Figure 3), while the electroretinogram showed normal results. Moreover, the optic nerves exhibited atrophic features, particularly in their intra-orbital segments, as shown in the orbito-cerebral MRI, and the biological work-up yielded negative findings.
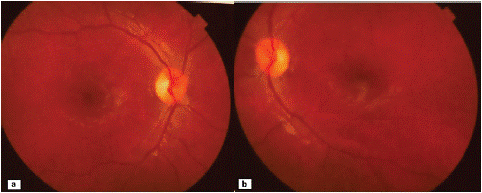
Figure 1: Retinography of the patient’s right (a) and left (b) eyes, showing a sectorial papillary pallor in the temporal region.
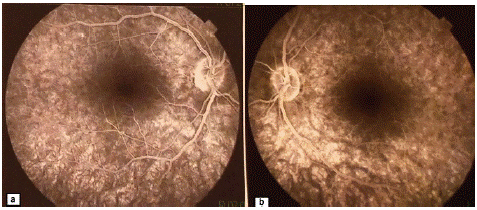
Figure 2: Fluorescein angiography of the right (a) and left (b) eye: no retention or leakage abnormalities in the late phase.
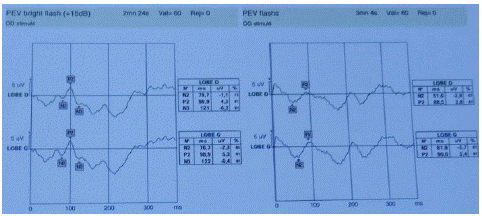
Figure 3: Fluorescein angiography of the right (a) and left (b) eye: no retention or leakage abnormalities in the late phase.
Leber's optic neuropathy was suspected, and a genetic study was conducted to investigate a possible mitochondrial mutation that could confirm the inheritance of the disease. A favorable result NC_012920.1:m.11778G>A was obtained six months later in the patient and his mother's ND4 mitochondrial gene, which confirmed the diagnosis of Leber's optic neuropathy.
After six months, the ophthalmological examination revealed residual visual acuity measurements of 1/60 on the Log Mar scale at a distance of 4m in the OD and PL+ in the OG, accompanied by total papillary pallor in both eyes (refer to figure 4). Since idebenone was not available, the patient received referral to low vision rehabilitation services, which offered functional rehabilitation exercises designed to stabilise and enhance suppletive fixation and facilitate combined ocular motor movements rehabilitation. The trial involved the use of mainly microscopic magnifying visual aids. An outdoor study was conducted to evaluate the effectiveness of using therapeutic chromatic selective lenses for managing problematic glare.
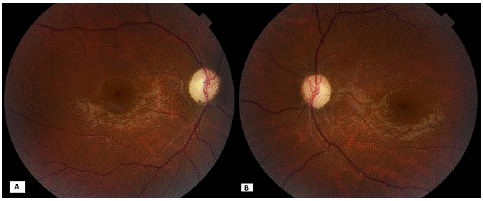
Figure 4: Retinography of the right eye (A) and left eye (B) 6 months after the onset of symptoms showing total papillary pallor.
After a five-month period of neofixation readjustment, the patient can now differentiate small objects and operate his smartphone as well as type texts on a small keyboard, tasks that previously proved to be extremely challenging. Additionally, he can differentiate fine details owing to the aid of his microscopic visual aid. Ergonomic neofixation was suggested and other equipment prescribed.
Discussion
Since the discovery of LHON, various therapeutic avenues have been tested, including Western medicine, traditional Chinese medicine, acupuncture, stem cells, gene therapy and various therapeutic molecules with different combinations of vitamins (B2, B3, B12, C, E and folic acid), ubiquinones, particularly idiquinone, gene therapy as well as various therapeutic molecules with different combinations of vitamins (B2, B3, B12, C, E and folic acid), ubiquinones including idebenone and other supplements (alpha-lipoic acid, carnitine, creatine, l-arginine, glutathione and dichloroacetate) [1,2]. Although promising research is underway, at present the main treatment for LHON remains supportive rather than curative, and relies mainly on visual rehabilitation and optical aids, which are very useful for people with central vision loss due to LHON [3].
Most people with LHON have permanent vision loss and are legally registered as visually impaired [4]. The 11778 mutation, with which our patient is affected, is characterised by the poorest outcome with only 4-17% visual recovery, with an average time to recovery of 36 months [5], with patients reporting that acute vision loss has a significant negative impact on their quality of life.
Vision and eye health problems have a considerable impact on general health and well-being, for example by reducing access to healthcare, increasing cardiovascular disease, limiting physical activity and aggravating depression and dementia.
Vision loss can be a distressing experience, particularly for older individuals (65 years and above) who have been found to experience psychological distress resulting from glaucoma, age-related macular degeneration, cataracts and diabetic retinopathy [6-12]. Furthermore, in adults over the age of 20, vision loss has been linked to an elevated risk of depression [6,7]. Nollett et al [10] estimated that 43% of young patients referred for low vision rehabilitation experienced clinically significant depression. This is on par with the prevalence of depression in cancer patients. It is important to note that these statistics represent real people who face common challenges in their daily lives, such as mobility, mental health, and social interaction.
Low vision rehabilitation is a feasible and promising option as most LHON patients are young adults with often intact peripheral vision; it aims to create a neo-fixation point and improve near activities. The goal of this therapy is to establish a new point of fixation and enhance near activities. However, it is crucial to note that low vision rehabilitation is comparable to any other form of rehabilitation and often necessitates a significant amount of time and effort before enhancing the patient's quality of life [13].
Psychological counselling is also recommended, ideally with a psychiatrist and/or specialist psychologist, to prevent phobic attitudes, depression and suicidal thoughts, and to help patients relearn effective strategies, regain self-confidence and accept the rehabilitation and compensatory measures that are essential for a return to independence and social life [14].
Conclusion
While LHON currently lacks a cure, individuals with LHON can maintain their work and social lives. It is crucial to evaluate the patient's functional vision and provide them with suitable supportive technology and personalised low vision care while considering their personality and surroundings. Emotional guidance and advice are fundamental to successful patient care.
Author Statements
Conflicts of Interest
The authors have no conflicts of interest to declare.
Funding
We have any financial sources.
Consent
Written informed consent was obtained from the patient and his parents for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
References
- Mashima Y, Hiida Y, Oguchi Y. Remission of Leber’s hereditary optic neuropathy with idebenone. Lancet. 1992;340(8815):368-9.
- Meyerson C, Van Stavern G, McClelland C. Leber hereditary optic neuropathy: current perspectives. Clin Ophthalmol. 2015;9:1165-76.
- Stelmack JA, Tang XC, Reda DJ, Rinne S, Mancil RM, Massof RW et al. Outcomes of the Veterans Affairs low vision intervention trial (LOVIT). Arch Ophthalmol. 2008;126(5):608-17.
- Kirkman MA, Korsten A, Leonhardt M, Dimitriadis K, De Coo IF, Klopstock T, et al. Quality of life in patients with Leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2009;50(7):3112-5.
- May-Yung Y, An-Guor W, Yau-Huei W. Leber’s hereditary optic neuropathy: A multifactorial diseaseProg Retin Eye Res. 2006;25(4):381-96.
- Cimarolli VR, Casten RJ, Rovner BW, Heyl V, Sörensen S, Horowitz A. Anxiety and depression in patients with advanced macular degeneration: current perspectives. Clin Ophthalmol. 2016;10:55-63.
- Varano M, Eter N, Winyard S, Wittrup-Jensen KU, Navarro R, Heraghty J. The emotional and physical impact of wet age-related macular degeneration: findings from the wAMD Patient and Caregiver Survey. Clin Ophthalmol. 2016;10:257-67.
- Yuzawa M, Fujita K, Tanaka E, Wang ECY. Assessing quality of life in the treatment of patients with age-related macular degeneration: clinical research findings and recommendations for clinical practice. Clin Ophthalmol. 2013;7:1325-32.
- Diniz-Filho A, Abe RY, Cho HJ, Baig S, Gracitelli CP, Medeiros FA. Fast visual field progression is associated with depressive symptoms in patients with glaucoma. Ophthalmology. 2016;123(4):754-9.
- Nollett CL, Bray N, Bunce C, Casten RJ, Edwards RT, Hegel MT, et al. High prevalence of untreated depression in patients accessing low-vision services. Ophthalmology. 2016;123(2):440-1.
- Evans JR, Fletcher AE, Wormald RP. Depression and anxiety in visu¬ally impaired older people. Ophthalmology. 2007;114(2):283-8.
- Brody BL, Gamst AC, Williams RA, Smith AR, Lau PW, Dolnak D, et al. Depression, visual acu¬ity, comorbidity, and disability associated with age-related macular degeneration. Ophthalmology. 2001;108(10):1893-900; discussion 1900-1.
- Wolffsohn JS, Cochrane AL. Design of the low vision quality-of-life questionnaire (LVQOL) and measuring the outcome of low-vision rehabilitation. Am J Ophthalmol. 2000;130(6):793-802.
- Zanlonghi X. La prise en charge des patients atteints de neuropathie optique héréditaire de Leber/ les associations de patients. J Fr Ophtalmol. 2022;45(8S1):S32-9. doi: 10.1016/S0181-5512(22)00448-X, PMID 36529476