
Abstract
Background: VKH disease is an autoimmune granulomatous panuveitis, primarily affecting young, pigmented women. A rare disease with a severe prognosis and a risk of ocular complications when diagnosed and treated late.
Methods: This is a retrospective descriptive study conducted in the department of internal medicine, between May 2016 and July 2023. We included five patients with VKH syndrome. The aim of our study was to determine the epidemiological, clinical, paraclinical, therapeutic and evolutionary characteristics of patients.
Results: All patients were Moroccan with an average age of 40 years and 5 months and a male predominance of 60%. The majority had an initial visual acuity between 3/10 and 4/10 and bilateral uveitis. 80% of patients were presented with extra-ocular signs. Patients received a corticosteroid bolus followed by oral corticosteroids, with only one patient requiring immunosuppressive therapy. A favorable evolution was noted.
Discussion: VKH disease is still considered a disease of people of Hispanic, Asian or Middle Eastern origin, with most patients between the second and fifth decade of life. It is a systemic disease with ocular, neurological, auditory and cutaneous involvement, although not all these symptoms are present in every patient. The mainstay of treatment is the early and intensive administration of systemic corticosteroids, and prognosis depends on rapid diagnosis and therapy.
Conclusion: VKH syndrome is a serious disease with severe complications for the eye, despite intensive treatment. Subclinical forms may underestimate its frequency. It is important to consider this diagnosis in all cases of bilateral uveitis.
Keywords: Vogt Koyanagi Harada; Granulomatous panuveitis; Retinal serous detachment; Corticosteroids
Introduction
Vogt-Koyanagi-Harada (VKH) disease is a rare autoimmune inflammatory disorder that affects several parts of the body, including the eyes, inner ear, meninges, skin and hair [1,2]. It mainly affects young, female subjects, such as Asians and Hispanics and is less common in Caucasians [3]. The exact mechanisms of VKH remain poorly understood.
Clinically, VKH presents with initial choroiditis, which progresses to granulomatous panuveitis with exudative retinal detachment. The auditory system may also be affected, and meningitis may occur [4,5,6]. The disease evolves in four phases: prodromal, acute, chronic convalescence and a recurrence phase [7,8].
Imaging techniques such as indocyanine green angiography (ICGA) and optical coherence tomography (OCT) have improved diagnostic accuracy and long-term follow-up. Treatment of VKH relies mainly on corticosteroids and immunomodulators but surgical options are available for complications such as subretinal fibrosis, glaucoma and cataracts. Disease progression and prognosis factors, including the stage of the disease at the time of treatment [9-12].
Our work includes both a retrospective study based on five cases of VKH treated in the Internal Medicine, and a comparative discussion with other case series from different origins.
Methods
This is a retrospective descriptive study of five patients, conducted over a period of 7years and 2 months, from May 2016 to July 2023. The study was carried out at the Internal Medicine B Department of the Mohammed V Military Teaching Hospital in Rabat. All cases referred for further etiological work-up of uveitis during this period were recruited for the study. All patients were of Moroccans.
In this study, we included patients with complete, incomplete or probable Vogt-Koyanagi-Harada syndrome. The diagnostic criteria used were the Revised Diagnostic Criteria (RDC), established by the International Committee on Nomenclature in 2001.
We excluded from our study patients with immunodeficiencyrelated uveitis, pseudo-uveitis, or pars planitis, as well as those with a history of ocular trauma or surgery, due to their distinct pathophysiological mechanisms.
Case 1
A 52-year-old patient with no previous medical history presented after six months of rapid progression of bilateral visual acuity loss, ocular redness and brow poliosis (Figure 1). Examination revealed granulomatous keratic precipitates, anterior chamber inflammation, iris atrophy and iridocapsular synechiae. Fundus examination revealed optic disc edema, weak macular reflex and serous retinal detachment. The etiological work-up, including viral, bacterial and immunological tests, was negative. Fluorescein angiography confirmed the fundus findings. On the basis of clinical and paraclinical criteria, a diagnosis of Vogt-Koyanagi-Harada (VKH) disease was made. The patient was treated with three monthly pulses of intravenous methylprednisolone, followed by a gradual reduction in prednisone and oral azathioprine. Uveitis regressed on treatment, but visual acuity stabilized at 3/10 before the patient was lost to follow-up.
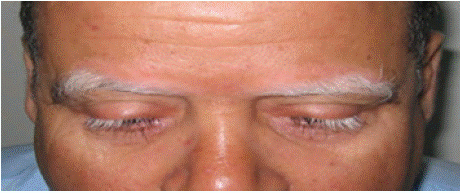
Figure 1: Bilateral lesions of eyebrow and eyelash poliosis.
Case 2
A 31-year-old woman with a one-year history of panuveitis under ophthalmologic follow-up presented with recently developed vitiligo lesions on the neck (Figure 2), evolving over three months, associated with poliosis of the scalp without alopecia, as well as complaints of hearing loss and vertigo. On examination, she was in good general condition.
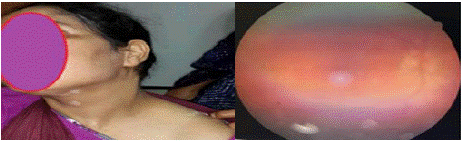
Figure 2: On the left, vitiligo lesions on the anterolateral aspect of the
patient’s neck. On the right, a retinal photograph of the eye, obscured by
cataracts, showing papilledema with DSR (delated segmental retinopathy)
and a “pinhead” appearance of the RPE (retinal pigment epithelium).
Ocular assessment showed bilateral visual acuity of 2/10, granulomatous keratic precipitates ("mutton-fat"), iridocapsular synechiae, anterior chamber inflammation (2+ Tyndall right eye, 3+ Tyndall left eye), and bilateral cataract. Fundus examination revealed a flat retina with good macular reflex, normal optic disc, perivasculitis, and serous retinal detachment in the right eye (Figure 2).
Audiometry confirmed sensorineural hearing loss. Extensive etiological workup including lumbar puncture, infectious serologies, and autoimmune testing was negative. The diagnosis of incomplete Vogt-Koyanagi-Harada (VKH) disease was considered. The patient received corticosteroid pulse therapy followed by oral and topical corticosteroids, with favorable clinical improvement.
Case 3
A 45-year-old male with an 18-month history of vitiligo presented with new-onset headaches, bilateral visual acuity loss, photophobia, and irritability. On physical examination, he was in good general health, but neurological assessment revealed pyramidal irritation, with brisk and diffuse deep tendon reflexes. Ophthalmologic examination revealed a visual acuity of 4/10 in the right eye and 3/10 in the left, with granulomatous keratic precipitates in both eyes, iridocapsular synechiae, and mild anterior chamber inflammation (1+ Tyndall right eye, 2+ Tyndall left eye). The lens was clear bilaterally. Fundus examination showed flat retinas with diffuse chorioretinal atrophy, optic disc edema, and multiple central and peripheral serous retinal detachments in both eyes. Dermatological evaluation revealed vitiligo lesions localized to the lumbar region, face, trunk, and all four limbs (Figure 3). Paraclinical investigations included cerebrospinal fluid analysis, which showed lymphocytic meningitis, and a normal brain MRI. HLA typing revealed the DRB1 genotype. The clinical presentation, coupled with ophthalmic, dermatologic, and neurological findings, led to the diagnosis of Vogt-Koyanagi- Harada (VKH) syndrome. The patient received a 5-day intravenous pulse of methylprednisolone (1 g/day), followed by oral corticosteroid therapy (1 mg/kg/day for 8 weeks, with gradual tapering). Visual acuity improved markedly within 48 hours of the pulse therapy, and the headaches resolved within one week of treatment.
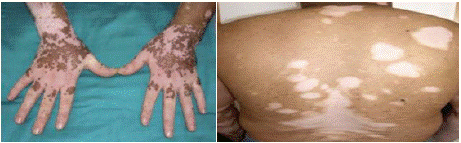
Figure 3: Vitiligo lesions on the patient’s hands and back.
Case 4
A 64-year-old male with a medical history of hypertension, atrial fibrillation, and gout presented with bilateral visual acuity loss, which had been progressively worsening over the past four months, along with ocular pain, dizziness, and tinnitus. On general examination, the patient was in good overall health. Ophthalmologic examination revealed visual acuity of 5/10 in both eyes, with granulomatous keratic precipitates ("mutton-fat") and iridocapsular synechiae bilaterally. The anterior chamber was deep and optically clear, with minimal inflammation (1+ Tyndall right eye and left eye). Both lenses were clear. Fundus examination showed a flat retina and multiple serous retinal detachments (SRD) in both eyes (Figure 4).
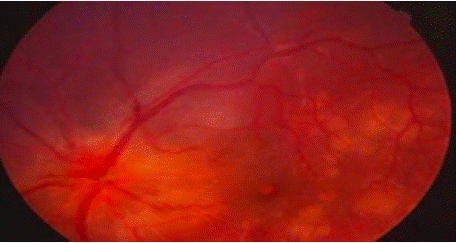
Figure 4: Retinal photograph of the left eye, highlighting stage 1
papilledema, multiple DSRs (dilated segmental retinopathies), and a large
multilobed DSR.
The patient was referred to internal medicine for an etiological workup. Dermatologic examination revealed vitiligo lesions on the face and both hands. A comprehensive diagnostic evaluation, including viral and bacterial serologies, immunological testing, and brain MRI, was negative, but HLA typing revealed an HLA DRB1 genotype. The combination of poliosis and uveitis led to a diagnosis of Vogt-Koyanagi-Harada (VKH) disease. The patient received a three-day pulse of intravenous methylprednisolone, followed by oral prednisone with a tapering schedule. Clinical improvement was noted, with regression of uveitis and improvement in visual acuity.
Case 5
A 10-year-old girl, followed for vitiligo on her hands (Figure 5) and trunk for the past 5 months, presented with bilateral visual acuity loss and photophobia that had been ongoing for 15 days. On physical examination, the patient was in good general health. Ophthalmologic examination revealed visual acuity of 5/10 in the right eye and 4/10 in the left, with granulomatous keratic precipitates ("mutton-fat") and iridocapsular synechiae bilaterally. The anterior chambers were deep and optically clear. Both lenses were transparent. Fundus examination showed a flat retina and good macular reflex, with a normal optic disc. Minimal vitreous haze was noted bilaterally, and serous retinal detachment (SRD) was present in both eyes (figure 5). No other abnormalities were found on the physical examination. Paraclinical investigations, including lumbar puncture, viral and bacterial serologies, and immunological testing, were all negative. The patient received a corticosteroid pulse followed by oral and topical corticosteroid therapy. Clinical improvement was noted, with good resolution of symptoms.
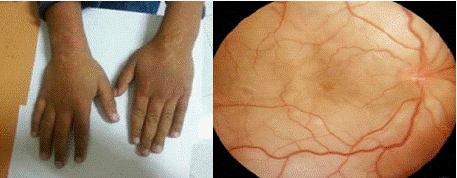
Figure 5: On the left, vitiligo lesions on the hands. On the right, retinal
photograph showing pallor of the optic fundus with DSR (dilated segmental
retinopathy).
Results
Table 1.
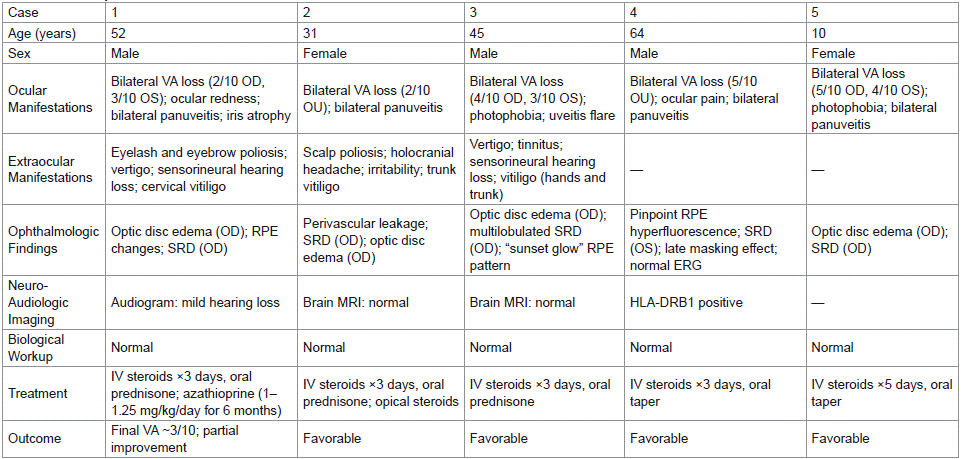
Table 1: Summary Table of Results.
Clinical Characteristics
This retrospective case series included five Moroccan patients diagnosed with Vogt-Koyanagi-Harada (VKH) syndrome, comprising three males (60%) and two females (40%), suggesting a slight male predominance. The mean age at diagnosis was 40.4 years (range: 10–64 years), with 80% of patients aged between 30 and 55 years. A majority (60%) resided in urban settings. No patient had a personal or familial history suggestive of autoimmune or uveitic disorders; only one had comorbid systemic conditions, namely hypertension, atrial fibrillation, and gout. All patients presented with bilateral visual acuity loss as the primary complaint, either isolated or associated with other neuro-ophthalmological symptoms. Photophobia and vertigo were each observed in 40% of cases, while ocular hyperemia, periocular pain, and holocranial headaches were reported in isolated instances.
Ocular involvement was bilateral in all cases (100%). At presentation, best-corrected visual acuity (BCVA) was =4/10 in 80% of eyes, with only 20% achieving BCVA =5/10. Slit-lamp biomicroscopy revealed anterior chamber inflammation with Tyndall phenomenon in 70% of eyes, poliosis of the eyelashes and eyebrows in 20%, and cataract formation in 20%. Iris atrophy and posterior synechiae were noted in 20% and 100% of cases, respectively. Fundoscopic examination demonstrated serous retinal detachment in 50% of eyes, optic disc edema in 30%, vitritis in 20%, and characteristic pigmentary changes of the retinal pigment epithelium (e.g., "sunset glow" fundus or pinpoint depigmentation) in 10%. Ocular motricity and pupillary light reflexes were preserved in all patients.
Extraocular manifestations were frequent: neurologic signs, including headache and signs of pyramidal tract irritation, were documented in one patient (20%); audio-vestibular symptoms, such as vertigo, tinnitus, and mild sensorineural hearing loss (confirmed by audiometry), were present in two patients (40%). Cutaneous manifestations were observed in four patients (80%), comprising vitiligo and poliosis of the scalp and eyelashes. Based on the 2001 revised diagnostic criteria for VKH, three patients fulfilled the criteria for the complete form of the disease, while two were classified as having the incomplete form.
Paraclinical Investigations
All five patients underwent multimodal ophthalmologic imaging. Fluorescein angiography (FA) was performed in all cases and demonstrated characteristic findings consistent with VKH disease. These included serous retinal detachments (SRDs) of varying morphology and extent, optic disc edema (stage 1), delayed choroidal perfusion, and multiple hyperfluorescent “pinpoint” foci indicative of choroidal inflammation. Notably, patient 1 exhibited bilateral SRDs with a large multilobulated detachment in the left eye; patient 2 showed signs of posterior uveitis with perivascular leakage and SRD; patient 3 had extensive pinpoint leakage and a large multilobulated SRD; patient 4 displayed delayed choroidal filling, punctate hyperfluorescence, and a large SRD with masking effect in late phases; and patient 5 presented with temporal pinpoint hyperfluorescence, discrete SRDs, and inflammatory nodular enhancement.
Optical coherence tomography (OCT) corroborated FA findings. All patients had evidence of subretinal fluid accumulation consistent with serous retinal detachment, with resolution observed following high-dose corticosteroid therapy. OCT follow-up demonstrated favorable anatomical response in all cases. In cases 3, 4, and 5, SRDs were unilateral and resolved under corticosteroid therapy without residual structural damage.
Extraocular investigations included brain MRI in two patients, which returned normal findings. HLA typing performed in two cases revealed the presence of HLA-DRB1 alleles. Audiometry confirmed mild sensorineural hearing loss in one patient, in line with auditory symptoms. Extensive infectious serologic screening for EBV, CMV, HSV, HBV, HCV, HIV, toxoplasmosis, and syphilis was negative in all patients. A full systemic work-up including CBC, ESR, CRP, liver and renal function tests, and chest radiography was conducted in all cases. Lumbar puncture in one patient revealed lymphocytic meningitis, confirming meningeal involvement.
Treatment
All patients received high-dose intravenous methylprednisolone (10 mg/kg/day), administered over 3 days in four cases and over 5 days in one. This was followed by oral prednisone at an initial dose of 1 mg/kg/day, tapered gradually over a period of 6 to 18 months, based on inflammatory activity. Topical corticosteroids were used adjunctively. Clinical improvement, particularly in terms of headache resolution, visual acuity recovery, and suppression of anterior chamber inflammation (Tyndall effect), was observed in all cases. One patient with severe posterior segment involvement refractory to corticosteroids was treated with azathioprine (1–1.25 mg/kg/day) for 6 months, achieving disease control.
Outcomes
Clinical evolution was favorable in all five patients. Resolution of uveitis, headaches, and vertigo was achieved in all cases. At final follow-up, visual acuity improved to =5/10 in 60% of affected eyes, while the remaining 40% retained a final visual acuity <5/10. Two patients experienced disease relapse during tapering of systemic corticosteroids, but no ocular complications were reported during follow-up in any patient.
Discussion
Epidemiological and Clinical Characteristics
Vogt-Koyanagi-Harada (VKH) disease demonstrates a distinct geographic and ethnic distribution, primarily affecting individuals with darker skin phototypes, particularly in East and Southeast Asia, the Middle East, and North Africa [13]. All patients in our series were North African. While VKH typically shows a slight female predominance, consistent with most global data, studies from Japan— including Sasamoto et al.—have reported a male predominance [14], which is mirrored in our cohort (60% male). The disease most commonly affects young adults aged 20 to 40 years. In our series, the mean age at onset was 40.4 years (range: 10–64), with one pediatric case that underscores the diagnostic complexity in children, where delayed presentation can obscure early and extraocular signs [15].
The diagnosis in all cases was clinical, supported by imaging (fluorescein angiography, brain MRI), lumbar puncture, and HLA typing to exclude differentials. Based on the revised 2001 VKH diagnostic criteria, our cohort showed a higher proportion of complete VKH (60%), which is likely attributable to late presentation, in contrast to the Japanese and European studies reporting a predominance of incomplete forms [15,16]. Visual disturbances were the chief complaint in all cases, although dermatologic or neurologic symptoms preceded ocular involvement in some patients. All patients had bilateral visual loss on presentation, consistent with the bilateral nature of VKH in over 90% of cases [13].
On ophthalmologic examination, all patients had a visual acuity of =5/10. Anterior uveitis was observed in all five patients, with one exhibiting vitritis. Posterior segment findings included optic disc edema (stage 1–2) in three patients and unilateral serous retinal detachment in all cases. Only one patient had early cataract formation. Fluorescein angiography confirmed disc edema in three cases, retinal detachment in all five, and drusen with macular changes in one. Optical coherence tomography (OCT), performed in one patient, was unremarkable.
Cutaneous involvement was present in 80% (vitiligo) and 40% (poliosis), closely aligning with literature that reports vitiligo and poliosis in 63% and 90% of chronic VKH cases, respectively [17]. The peri-limbal vitiligo (Sugiura’s sign), common in Asian populations [16], was absent in our patients. Neurologically, cerebrospinal fluid (CSF) analysis was normal in most patients except one with lymphocytic meningitis; this is consistent with literature indicating CSF pleocytosis in the early inflammatory phase [16,17]. Auditory symptoms were reported in two patients (40%), including vertigo and dysacusis, in line with studies noting auditory dysfunction in up to 74% of VKH patients, which may resolve over time [17].
Treatment and Outcomes
A review of recent VKH cohorts across various countries reveals significant heterogeneity in disease evolution, relapse rates, and visual outcomes—largely influenced by treatment strategies, timing, and access to immunosuppressive therapy.
In most studies, including those from Iran (2006) and Brazil (2020), high rates of chronic VKH (up to 90%) and poor visual prognosis were reported, despite corticosteroid use. These poor outcomes may be attributed to delayed treatment initiation, insufficient duration of immunosuppression, or inadequate disease monitoring. The Brazilian cohort, although treated with multiple immunosuppressants including biologics, showed that all patients were already in chronic stage at diagnosis, which limits treatment efficacy.
In India (Tamil Nadu, 2010) and the USA (2020), relapse rates were also notable (up to 36.4%), with visual acuity <20/200 in 13–21% of eyes, suggesting that steroid therapy alone may be insufficient in preventing recurrence without robust immunomodulatory control.
More recent studies, such as those from Saudi Arabia (2022) and India (Andhra Pradesh, 2022), highlight improved outcomes, possibly due to earlier intervention and broader use of immunosuppressants like mycophenolate mofetil (MMF), azathioprine (AZA), and methotrexate (MTX). However, recurrence remained frequent (44– 47%), and visual impairment <20/40 was still observed in over onethird of eyes [17].
In contrast, our cohort demonstrated a clearly more favorable profile, with:
• No cases of chronic VKH or SGF,
• Low relapse rate, limited to a single patient,
• Good visual recovery, with only one case of persistent poor VA (3/10),
• Minimal ocular complications, with only one early cataract.
These results likely stem from the early initiation of high-dose IV corticosteroids, prolonged and controlled tapering, and prompt escalation to immunosuppressants in refractory cases. Our experience underscores the critical importance of timing, aggressiveness, and consistency of treatment in altering the natural course of VKH.
Conclusion
Vogt-Koyanagi-Harada (VKH) disease is a bilateral granulomatous panuveitis often associated with systemic manifestations such as meningismus, vitiligo, alopecia, and auditory symptoms. Diagnosis relies on clinical presentation and imaging tools like Spectral-Domain Optical Coherence Tomography (SD-OCT), fluorescein angiography, and B-scan. The 2001 revised criteria classify VKH into complete, incomplete, and probable forms. High-dose systemic corticosteroids remain the cornerstone of treatment, with immunosuppressants reserved for severe or refractory cases. Early, aggressive therapy generally yields a good visual prognosis.
References
- Patil YB, Garg R, Rajguru JP, Sirsalmath M, Bevinakatti VA, Kumar M, et al. Vogt-Koyanagi-Harada (VKH) syndrome: A new perspective for healthcare professionals. J Family Med Prim Care. 2020; 9: 31-35.
- Stern EM, Nataneli N. Vogt-Koyanagi-Harada Syndrome. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023.
- Lavezzo MM, Sakata VM, Morita C, Rodriguez EE, Abdallah SF, da Silva FT, et al. Vogt-Koyanagi-Harada disease: review of a rare autoimmune disease targeting antigens of melanocytes. Orphanet J Rare Dis. 2016; 11: 29.
- Hirooka K, Saito W, Namba K, Mizuuchi K, Iwata D, Hashimoto Y, et al. Significant role of the choroidal outer layer during recovery from choroidal thickening in Vogt-Koyanagi-Harada disease patients treated with systemic corticosteroids. BMC Ophthalmol. 2015; 15: 181.
- Nakamura S, Nakazawa M, Yoshioka M, Nagano I, Nakamura H, Onodera J, et al. Melanin-laden macrophages in cerebrospinal fluid in Vogt-Koyanagi- Harada syndrome. Arch Ophthalmol. 1996; 114: 1184–1188.
- Fang W, Yang P. Vogt-Koyanagi-Harada syndrome. Curr Eye Res. 2008; 33: 517–523.
- Lodhi SAK, Reddy JML, Peram V. Clinical spectrum and management options in Vogt-Koyanagi-Harada disease. Clin Ophthalmol. 2017; 11: 1399–1406.
- Urzua CA, Herbort CP Jr, Valenzuela RA, Abu El-Asrar AM, Arellanes-Garcia L, Schlaen A, et al. Initial-onset acute and chronic stages are two distinctive courses of Vogt-Koyanagi-Harada disease. J Ophthalmic Inflamm Infect. 2020; 10: 23.
- Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001; 131: 647–652.
- Hedayatfar A, Khochtali S, Khairallah M, Takeuchi M, El-Asrar AA, Herbort CP Jr. “Revised diagnostic criteria” for Vogt-Koyanagi-Harada disease fail to improve disease management. J Curr Ophthalmol. 2018; 31: 1–7.
- Bouchenaki N, Herbort CP. The contribution of indocyanine green angiography to the appraisal and management of Vogt-Koyanagi-Harada disease. Ophthalmology. 2001; 108: 54–64.
- Nakai K, Gomi F, Ikuno Y, Yasuno Y, Nouchi T, Ohguro N, et al. Choroidal observations in Vogt-Koyanagi-Harada disease. 2012; 250: 1089-1095.
- Du L, Kijlstra A, Yang P. Vogt-Koyanagi-Harada disease: novel insights into pathophysiology, diagnosis and treatment. 2016.
- Smith ME, Kincaid MC, West CE. Anatomie et réfraction. 2004.
- Damico FM, et al. New insights into Vogt-Koyanagi-Harada disease. Arq Bras Oftalmol. 2009; 72: 413-420.
- El Houari G, Rachid R, Boukhrissa M, Khtibari Z, Karami R, Baha W, et al. Service d’ophtalmologie Adulte - Casablanca. Journal de la Société Marocaine d’Ophtalmologie. 2015.
- Accortini M. Uveitis Webinar 23 (7 Jan 2023): Vogt Koyanagi Harada Disease. 2024.